In Business Since 2004
Relevant Publications from Cassia Personnel
Complete List of Canonical Post-transcriptional Modifications in the Bacillus subtilis Ribosome and their Link to RbgA Driven Large Subunit Assembly
Nucleic Acids Res. 2024, 52, 11203-11217.

Abstract: Ribosomal RNA modifications in prokaryotes have been sporadically studied, but there is a lack of a comprehensive picture of modification sites across bacterial phylogeny. Bacillus subtilis is a preeminent model organism for gram-positive bacteria, with a well-annotated and editable genome, convenient for fundamental studies and industrial use. Yet remarkably, there has been no complete characterization of its rRNA modification inventory. By expanding modern MS tools for the discovery of RNA modifications, we found a total of 25 modification sites in 16S and 23S rRNA of B. subtilis, including the chemical identity of the modified nucleosides and their precise sequence location. Furthermore, by perturbing large subunit biogenesis using depletion of an essential factor rbgA and measuring the completion of 23S modifications in the accumulated intermediate, we provide a first look at the order of modification steps during the late stages of assembly in B. subtilis. While our work expands the knowledge of bacterial rRNA modification patterns, adding B. subtilis to the list of fully annotated species after Escherichia coli and Thermus thermophilus, in a broader context, it provides the experimental framework for discovery and functional profiling of rRNA modifications to ultimately elucidate their role in ribosome biogenesis and translation.
Pre-B Cell Receptor Acts as a Selectivity Switch for Galectin-1 at the Pre-B Cell Surface
Cell Reports 2024, 43, 114541.
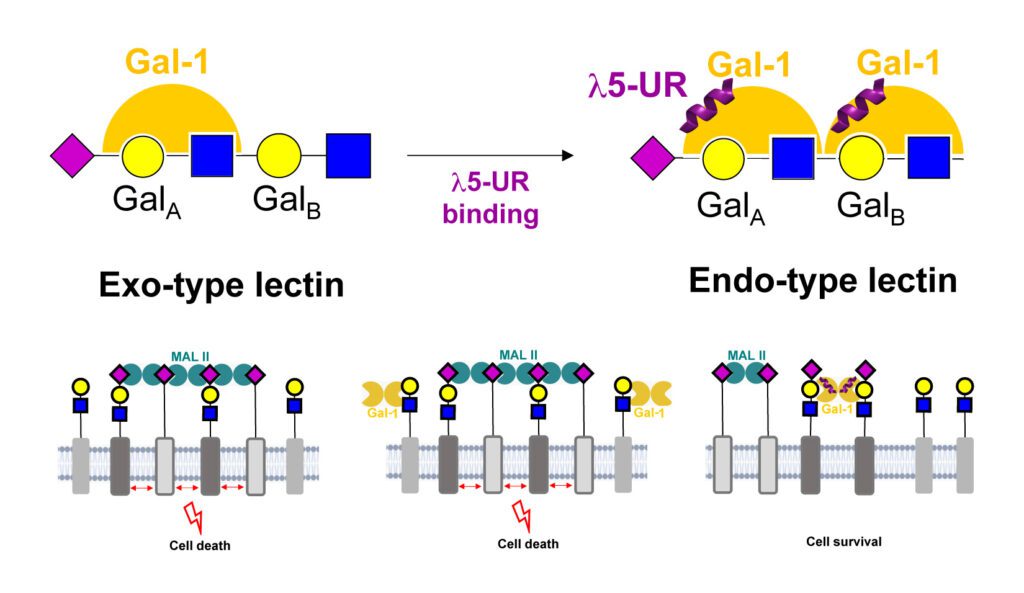
Abstract: Galectins are glycan-binding proteins translating the sugar-encoded information of cellular glycoconjugates into physiological activities, including immunity, cell migration, and signaling. Galectins also interact with non-glycosylated partners in the extracellular milieu, among which the pre-B cell receptor (pre-BCR) during B cell development. How these interactions might interplay with the glycan-decoding function of galectins is unknown. Here, we perform NMR experiments on native membranes to monitor Gal-1 binding to physiological cell surface ligands. We show that pre-BCR interaction changes Gal-1 binding to glycosylated pre-B cell surface receptors. At the molecular and cellular levels, we identify α-2,3-sialylated motifs as key targeted epitopes. This targeting occurs through a selectivity switch increasing Gal-1 contacts with α-2,3-sialylated poly-N-acetyllactosamine upon pre-BCR interaction. Importantly, we observe that this switch is involved in the regulation of pre-BCR activation. Altogether, this study demonstrates that interactions to non-glycosylated proteins regulate the glycan-decoding functions of galectins at the cell surface.
Tandem Mass Spectrometry Across Platforms
Analytical Chem. 2024, 96, 5478-5488.

Abstract: PubChem serves as a comprehensive repository, housing over 100 million unique chemical structures representing the breadth of our chemical knowledge across numerous fields including metabolism, pharmaceuticals, toxicology, cosmetics, agriculture, and many more. Rapid identification of these small molecules increasingly relies on electrospray ionization (ESI) paired with tandem mass spectrometry (MS/MS), particularly by comparison to genuine standard MS/MS data sets. Despite its widespread application, achieving consistency in MS/MS data across various analytical platforms remains an unaddressed concern. This study evaluated MS/MS data derived from one hundred molecular standards utilizing instruments from five manufacturers, inclusive of quadrupole time-of-flight (QTOF) and quadrupole orbitrap “exactive” (QE) mass spectrometers by Agilent (QTOF), Bruker (QTOF), SCIEX (QTOF), Waters (QTOF), and Thermo QE. We assessed fragment ion variations at multiple collisional energies (0, 10, 20, and 40 eV) using the cosine scoring algorithm for comparisons and the number of fragments observed. A parallel visual analysis of the MS/MS spectra across instruments was conducted, consistent with a standard procedure that is used to circumvent the still prevalent issue of mischaracterizations as shown for dimethyl sphingosine and C20 sphingosine. Our analysis revealed a notable consistency in MS/MS data and identifications, with fragment ions’ m/z values exhibiting the highest concordance between instrument platforms at 20 eV, the other collisional energies (0, 10, and 40 eV) were significantly lower. While moving toward a standardized ESI MS/MS protocol is required for dependable molecular characterization, our results also underscore the continued importance of corroborating MS/MS data against standards to ensure accurate identifications. Our findings suggest that ESI MS/MS manufacturers, akin to the established norms for gas chromatography mass spectrometry instruments, should standardize the collision energy at 20 eV across different instrument platforms.
Pytheas: A Software Package for the Automated Analysis of RNA Sequences and Modifications via Tandem Mass Spectrometry
Nat. Commun. 2022, 13, 2424-2436.
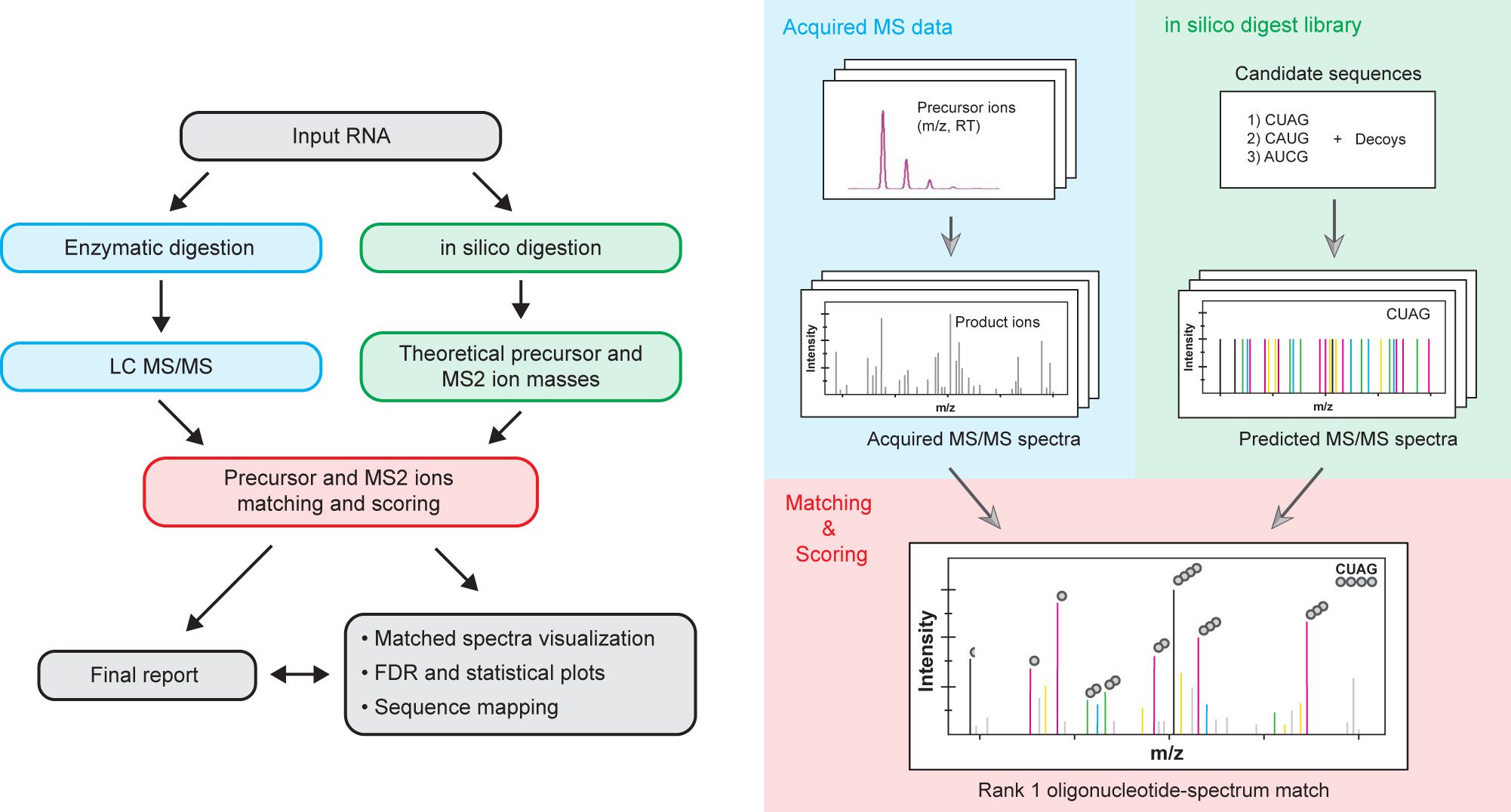
Abstract: Mass spectrometry is an important method for analysis of modified nucleosides ubiquitously present in cellular RNAs, in particular for ribosomal and transfer RNAs that play crucial roles in mRNA translation and decoding. Furthermore, modifications have effect on the lifetimes of nucleic acids in plasma and cells and are consequently incorporated into RNA therapeutics. To provide an analytical tool for sequence characterization of modified RNAs, we developed Pytheas, an open-source software package for automated analysis of tandem MS data for RNA. The main features of Pytheas are flexible handling of isotope labeling and RNA modifications, with false discovery rate statistical validation based on sequence decoys. We demonstrate bottom-up mass spectrometry characterization of diverse RNA sequences, with broad applications in the biology of stable RNAs, and quality control of RNA therapeutics and mRNA vaccines.
Mechanism of Rate Acceleration of Radical C-C Bond Formation Reaction by a Radical SAM GTP 3′,8-Cyclase
J. Am. Chem. Soc. 2020, 142, 9314-9326.
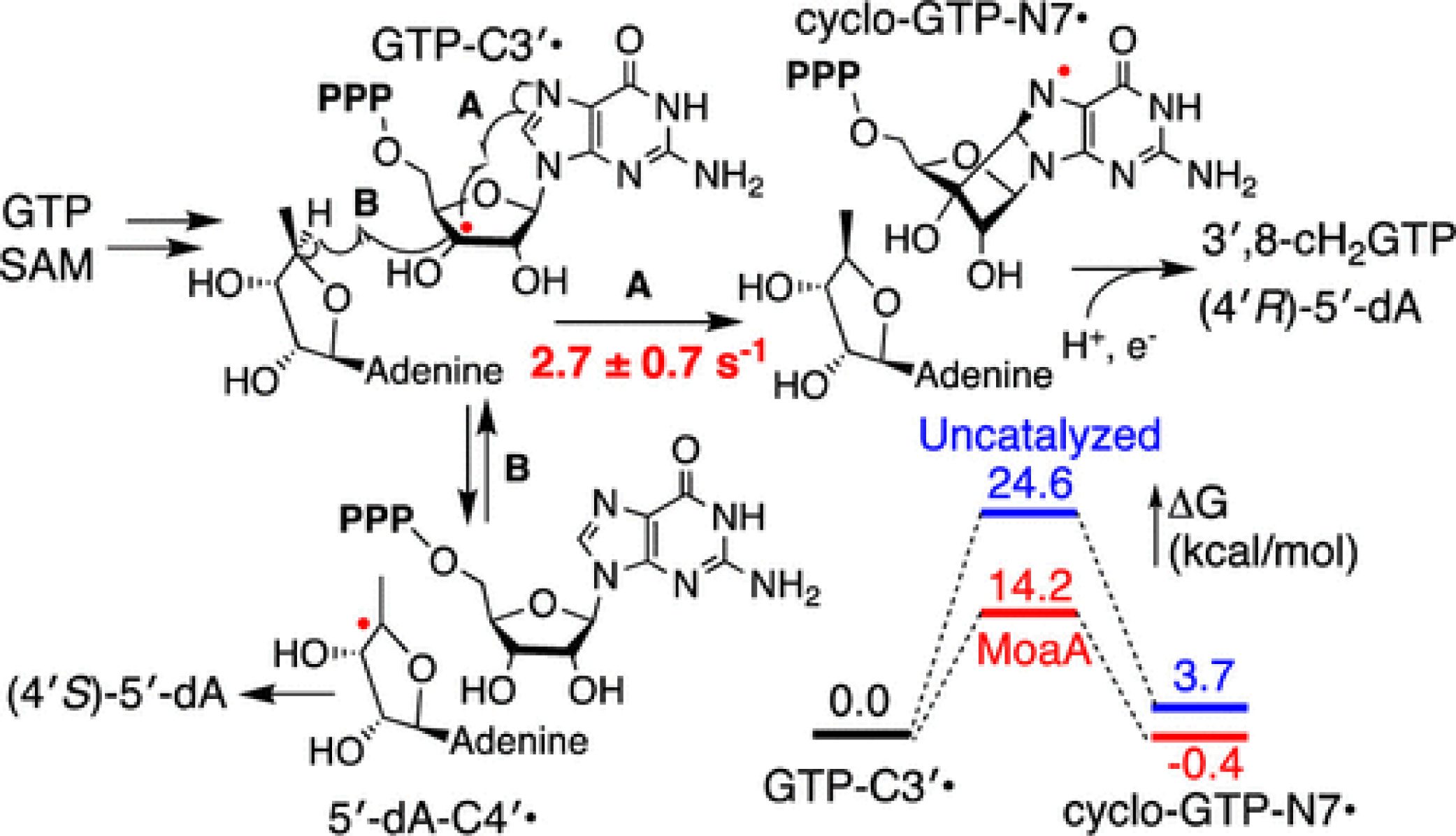
Abstract: While the number of characterized radical S-adenosyl-L-methionine (SAM) enzymes is increasing, the roles of these enzymes in radical catalysis remain largely ambiguous. In radical SAM enzymes, the slow radical initiation step kinetically masks the subsequent steps, making it impossible to study the kinetics of radical chemistry. Due to this kinetic masking, it is unknown whether the subsequent radical reactions require rate acceleration by the enzyme active site. Here, we report the first evidence that a radical SAM enzyme MoaA accelerates the radical-mediated C-C bond formation. MoaA catalyzes an unprecedented 3′,8-cyclization of GTP into 3′,8-cyclo-7,8-dihydro-GTP (3′,8-cH2GTP) during the molybdenum cofactor (Moco) biosynthesis. Through a series of EPR and biochemical characterizations, we found that MoaA catalyzes a shunt pathway in which an on-pathway intermediate, GTP C-3′ radical, abstracts H-4′ atom from (4′R)-5′-deoxyadenosine (5′-dA) to transiently generate 5′-deoxyadenos-4′-yl radical (5′-dA-C4′•) that is subsequently reduced stereospecifically to yield (4′S)-5′-dA. Detailed kinetic characterization of the shunt and the main pathways provided the comprehensive view of MoaA kinetics and determined the rate of the on-pathway 3′,8-cyclization step as 2.7 ± 0.7 s–1. Together with DFT calculations, this observation suggested that the 3′,8-cyclization by MoaA is accelerated by 6-9 orders of magnitude. Further experimental and theoretical characterizations suggested that the rate acceleration is achieved mainly by constraining the triphosphate and guanine base positions while leaving the ribose flexible, and a transition state stabilization through H-bond and electrostatic interactions with the positively charged R17 residue. This is the first evidence for rate acceleration of radical reactions by a radical SAM enzyme and provides insights into the mechanism by which radical SAM enzymes accelerate radical chemistry.
Paradigm Shift for Radical S-Adenosyl-L-Methionine Reactions: The Organometallic Intermediate Ω Is Central to Catalysis
J. Am. Chem. Soc. 2018, 140, 8634-8638.
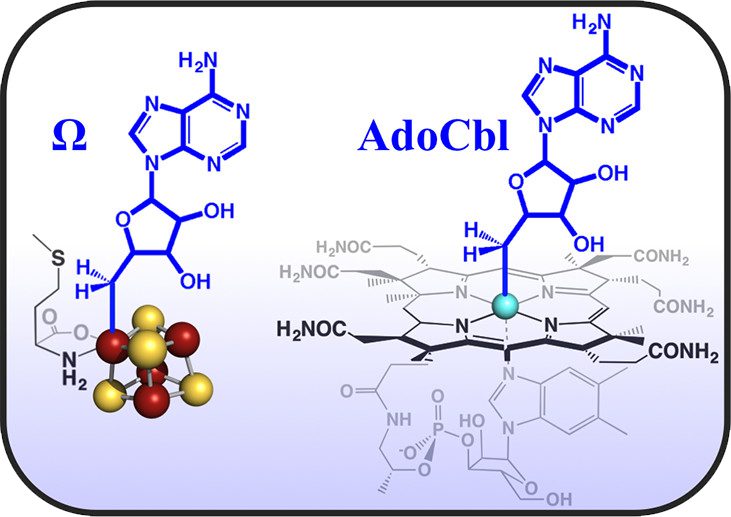
Abstract: Radical S-adenosyl-L-methionine (SAM) enzymes comprise a vast superfamily catalyzing diverse reactions essential to all life through homolytic SAM cleavage to liberate the highly reactive 5′-deoxyadenosyl radical (5′-dAdo•). Our recent observation of a catalytically competent organometallic intermediate Ω that forms during reaction of the radical SAM (RS) enzyme pyruvate formate-lyase activating-enzyme (PFL-AE) was therefore quite surprising, and led to the question of its broad relevance in the superfamily. We now show that Ω in PFL-AE forms as an intermediate under a variety of mixing order conditions, suggesting it is central to catalysis in this enzyme. We further demonstrate that Ω forms in a suite of RS enzymes chosen to span the totality of superfamily reaction types, implicating Ω as essential in catalysis across the RS superfamily. Finally, EPR and electron nuclear double resonance spectroscopy establish that Ω involves an Fe-C5′ bond between 5′-dAdo• and the [4Fe-4S] cluster. An analogous organometallic bond is found in the well-known adenosylcobalamin (coenzyme B12) cofactor used to initiate radical reactions via a 5′-dAdo• intermediate. Liberation of a reactive 5′-dAdo• intermediate via homolytic metal-carbon bond cleavage thus appears to be similar for Ω and coenzyme B12. However, coenzyme B12 is involved in enzymes catalyzing only a small number (~12) of distinct reactions, whereas the RS superfamily has more than 100 000 distinct sequences and over 80 reaction types characterized to date. The appearance of Ω across the RS superfamily therefore dramatically enlarges the sphere of bio-organometallic chemistry in nature.
19F-Site-Specific-Labeled Nucleotides for Nucleic Acid Structural Analysis by NMR
Methods Enzymol. 2016, 556, 59-87.
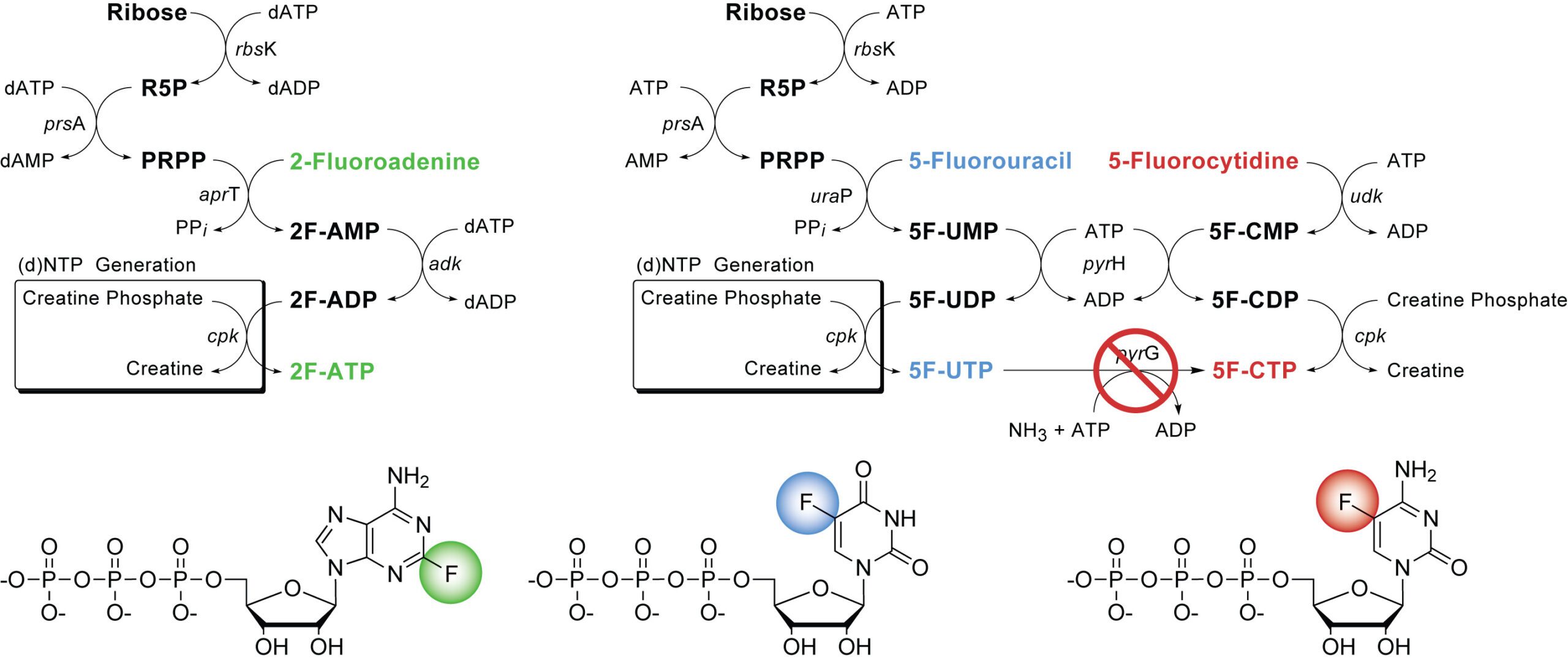
Abstract: Naturally occurring RNA lacks fluorine-19 (19F), thus, their specifically fluorinated counterparts are particularly well suited to noninvasively monitoring the dynamic conformational properties and ligand-binding interactions of the RNA. For nuclear magnetic resonance (NMR) spectroscopy, 19F-NMR of fluorine-substituted RNA provides an attractive, site-specific probe for structure determination in solution. Advantages of 19F include high NMR sensitivity (83% of 1H), high natural abundance (100%), and the extreme sensitivity of 19F to the chemical environment leading to a large range of chemical shifts. The preparation of base-substituted 2-fluoropurine and 5-fluoropyrimidine 5′-triphosphates (2F-ATP/5F-CTP/5F-UTP) can be carried out using efficient enzymatic synthesis methods. Both pyrimidine analogs, 5-fluorouridine and 5-fluorocytidine, as well as, 2-fluoroadenosine are readily incorporated into RNA transcribed in vitro using T7 RNA polymerase.
Quantitative Analysis of rRNA Modifications Using Stable Isotope Labeling and Mass Spectrometry
J. Am. Chem. Soc. 2014, 136, 2058-2069.
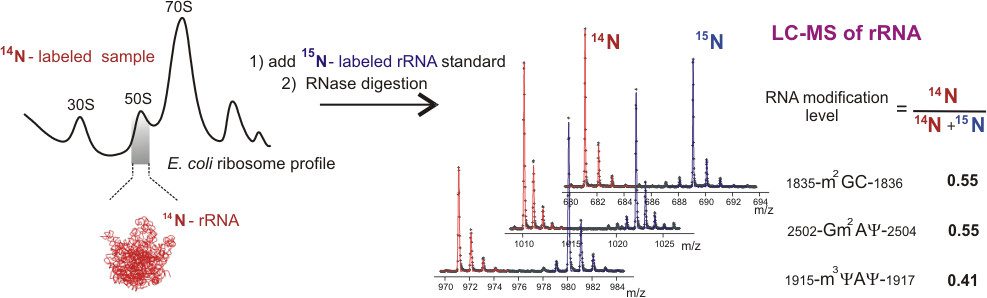
Abstract: Post-transcriptional RNA modifications that are introduced during the multistep ribosome biogenesis process are essential for protein synthesis. The current lack of a comprehensive method for a fast quantitative analysis of rRNA modifications significantly limits our understanding of how individual modification steps are coordinated during biogenesis inside the cell. Here, an LC-MS approach has been developed and successfully applied for quantitative monitoring of 29 out of 36 modified residues in the 16S and 23S rRNA from Escherichia coli. An isotope labeling strategy is described for efficient identification of ribose and base methylations, and a novel metabolic labeling approach is presented to allow identification of MS-silent pseudouridine modifications. The method was used to measure relative abundances of modified residues in incomplete ribosomal subunits compared to a mature (15)N-labeled rRNA standard, and a number of modifications in both 16S and 23S rRNA were present in substoichiometric amounts in the preribosomal particles. The RNA modification levels correlate well with previously obtained profiles for the ribosomal proteins, suggesting that RNA is modified in a schedule comparable to the association of the ribosomal proteins. Importantly, this study establishes an efficient workflow for a global monitoring of ribosomal modifications that will contribute to a better understanding of mechanisms of RNA modifications and their impact on intracellular processes in the future.
Characterization of the Ribosome Biogenesis Landscape in E. coli Using Quantitative Mass Spectrometry
J. Mol. Biol. 2013, 425, 767-779.
Abstract: The ribosome is an essential and highly complex biological system in all living cells. A large body of literature on the assembly of the ribosome in vitro is available, but a clear picture of this process inside the cell has yet to emerge. Here, we directly characterized in vivo ribosome assembly intermediates and associated assembly factors from wild-type Escherichia coli cells using a general quantitative mass spectrometry (qMS) approach. The presence of distinct populations of ribosome assembly intermediates was verified using an in vivo stable isotope pulse-labeling approach, and their exact ribosomal protein contents were characterized against an isotopically labeled standard. The model-free clustering analysis of the resultant protein levels for the different ribosomal particles produced four 30S assembly groups that correlate very well with previous in vitro assembly studies of the small ribosomal subunit and six 50S assembly groups that clearly define an in vivo assembly landscape for the larger ribosomal subunit. In addition, de novo proteomics identified a total of 21 known and potentially new ribosome assembly factors co-localized with various ribosomal particles. These results represent new in vivo assembly maps of the E. coli 30S and 50S subunits, and the general qMS approach should prove to be a solid platform for future studies of ribosome biogenesis across a host of model organisms.
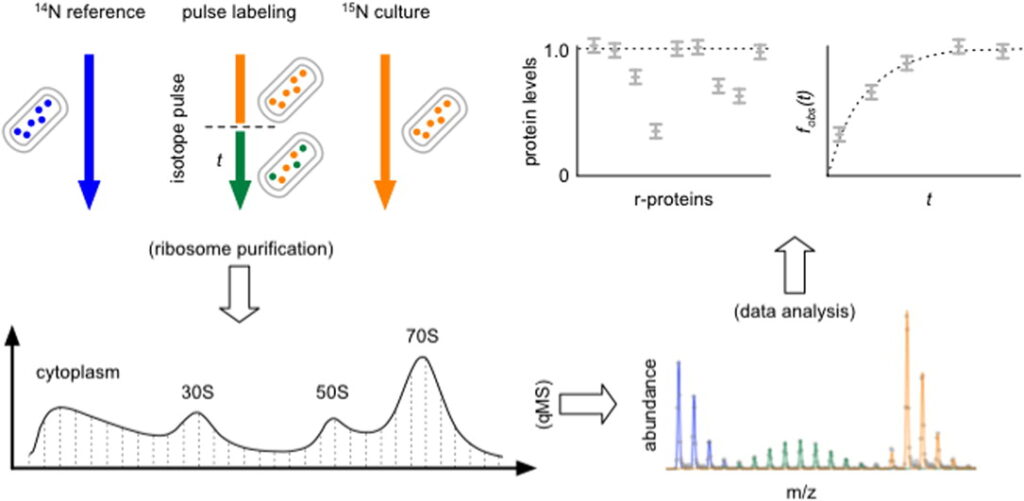
Measuring the Dynamics of E. coli Ribosome Biogenesis Using Pulse-Labeling and Quantitative Mass Spectrometry
Mol. Biosyst. 2012, 8, 3325-3334.
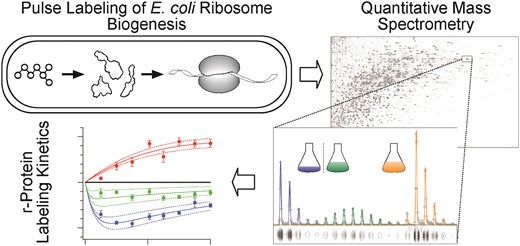
Abstract: The ribosome is an essential organelle responsible for cellular protein synthesis. Until recently, the study of ribosome assembly has been largely limited to in vitro assays, with few attempts to reconcile these results with the more complex in vivo ribosome biogenesis process. Here, we characterize the ribosome synthesis and assembly pathway for each E. coli ribosomal protein (r-protein) in vivo using a stable isotope pulse-labeling timecourse. Isotope incorporation into assembled ribosomes was measured by quantitative mass spectrometry (qMS) and fit using steady-state flux models. Most r-proteins exhibit precursor pools ranging in size from 0% to 7% of completed ribosomes, and that the sizes of these individual r-protein pools correlate well with the order of r-protein binding in vitro. Additionally, we observe anomalously large precursor pools for specific r-proteins with known extra-ribosomal functions and we have detected three r-proteins with significant turnover during steady-state growth. Taken together, this highly precise, time-dependent proteomic qMS approach should prove useful in future studies of ribosome biogenesis and could be easily extended to explore other complex biological processes in a cellular context.
An Active-Site Guanine Participates in glmS Ribozyme Catalysis in Its Protonated State
J. Am. Chem. Soc. 2011, 133, 18388-18396.
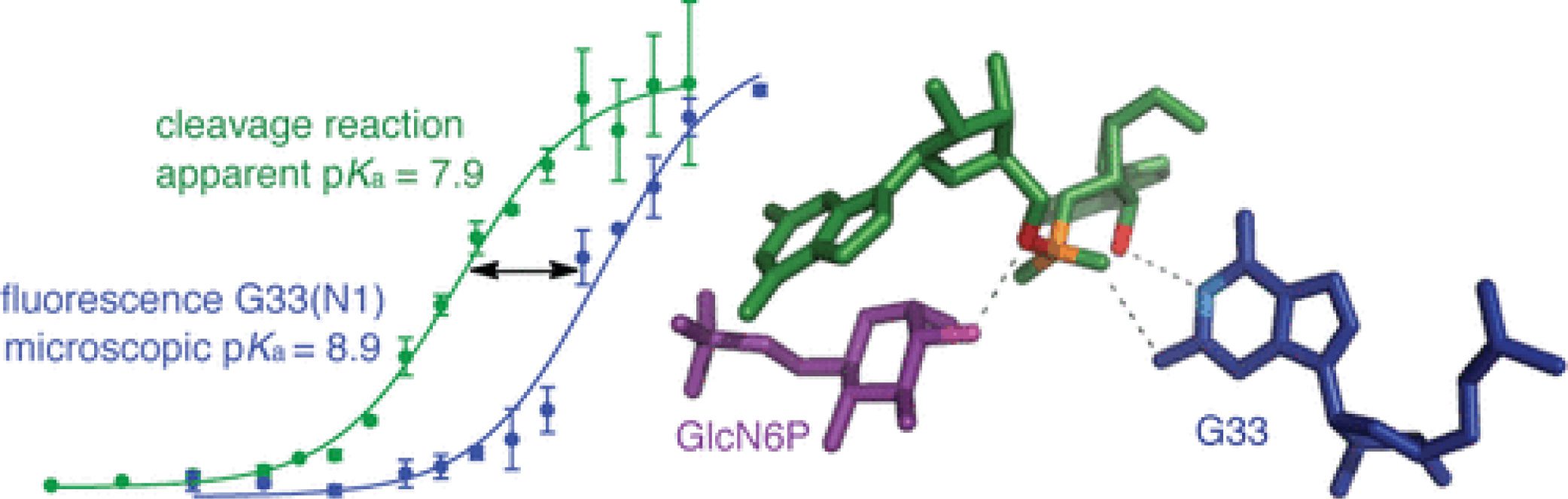
Abstract: Active-site guanines that occupy similar positions have been proposed to serve as general base catalysts in hammerhead, hairpin, and glmS ribozymes, but no specific roles for these guanines have been demonstrated conclusively. Structural studies place G33(N1) of the glmS ribozyme of Bacillus anthracis within hydrogen-bonding distance of the 2′-OH nucleophile. Apparent pKa values determined from the pH dependence of cleavage kinetics for wild-type and mutant glmS ribozymes do not support a role for G33, or any other active-site guanine, in general base catalysis. Furthermore, discrepancies between apparent pKa values obtained from functional assays and microscopic pKa values obtained from pH–fluorescence profiles with ribozymes containing a fluorescent guanosine analogue, 8-azaguanosine, at position 33 suggest that the pH-dependent step in catalysis does not involve G33 deprotonation. These results point to an alternative model in which G33(N1) in its neutral, protonated form donates a hydrogen bond to stabilize the transition state.
Identification of 5-Hydroxycytidine at Position 2501 Concludes Characterization of Modified Nucleotides in E. coli 23S rRNA
J. Mol. Biol. 2011, 411, 529-536.
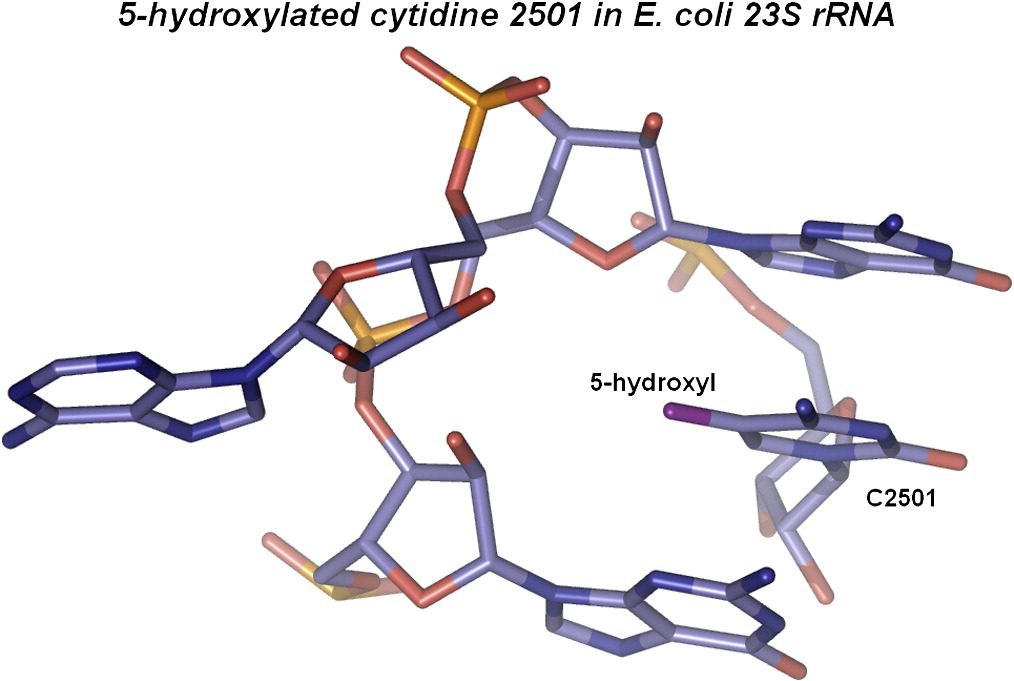
Abstract: Complete characterization of a biomolecule’s chemical structure is crucial in the full understanding of the relations between their structure and function. The dominating components in ribosomes are ribosomal RNAs (rRNAs), and the entire rRNA – but a single modified nucleoside at position 2501 in 23S rRNA – has previously been characterized in the bacterium Escherichia coli. Despite a first report nearly 20 years ago, the chemical nature of the modification at position 2501 has remained elusive, and attempts to isolate it have so far been unsuccessful. We unambiguously identify this last unknown modification as 5-hydroxycytidine – a novel modification in RNA. Identification of 5-hydroxycytidine was completed by liquid chromatography under nonoxidizing conditions using a graphitized carbon stationary phase in combination with ion trap tandem mass spectrometry and by comparing the fragmentation behavior of the natural nucleoside with that of a chemically synthesized ditto. Furthermore, we show that 5-hydroxycytidine is also present in the equivalent position of 23S rRNA from the bacterium Deinococcus radiodurans. Given the unstable nature of 5-hydroxycytidine, this modification might be found in other RNAs when applying the proper analytical conditions as described here.
A Nano-Chip-LC/MSn Based Strategy for Characterization of Modified Nucleosides Using Reduced Porous Graphitic Carbon as a Stationary Phase
J. Am. Soc. Mass Spectrom. 2011, 22, 1242-1251.
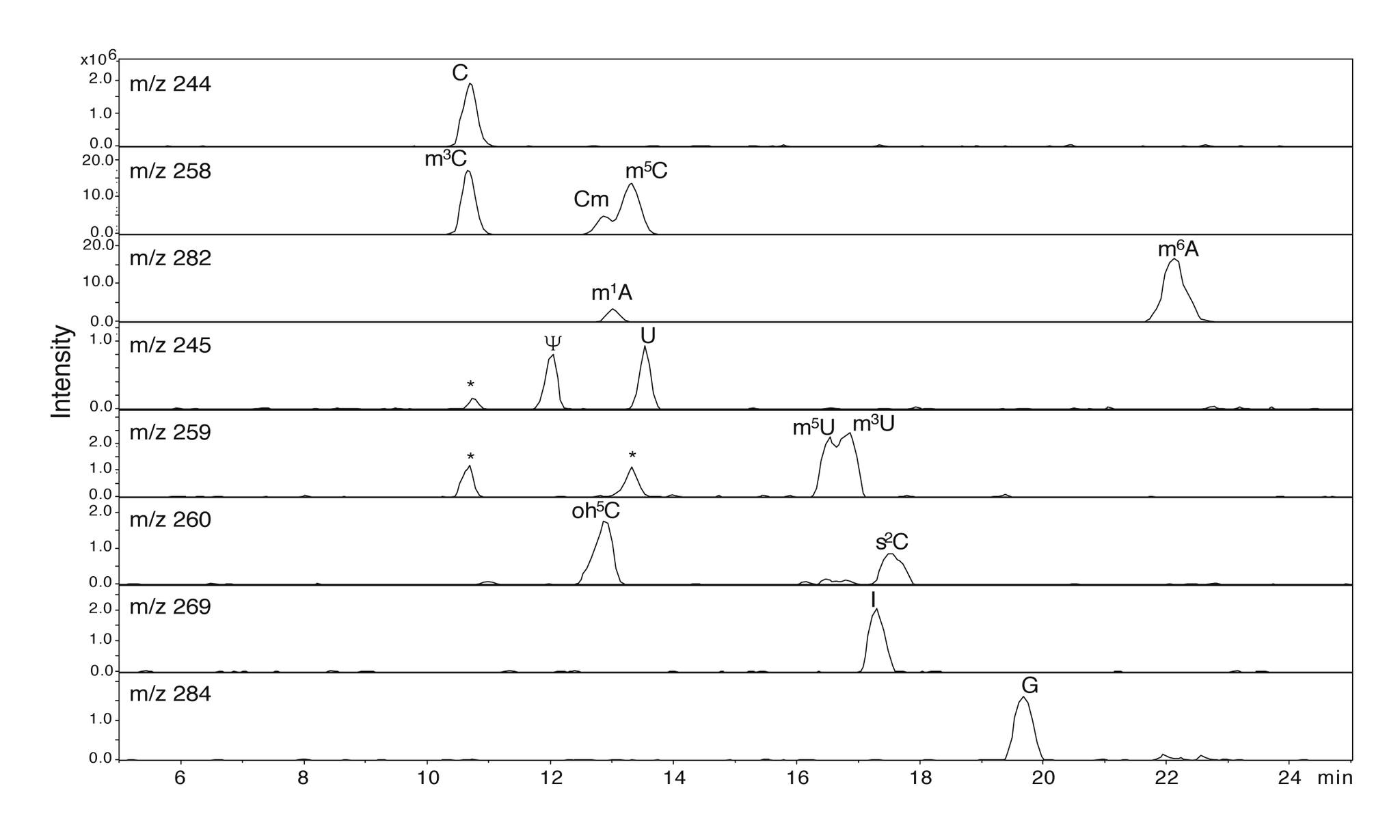
Abstract: LC/MS analysis of ribonucleosides is traditionally performed by reverse phase chromatography on silica based C18 type stationary phases using MS compatible buffers and methanol or acetonitrile gradients. Due to the hydrophilic and polar nature of nucleosides, down-scaling C18 analytical methods to a two-column nano-flow setup is inherently difficult. We present a nano-chip LC/MS ion-trap strategy for routine characterization of RNA nucleosides in the fmol range. Nucleosides were analyzed in positive ion mode by reverse phase chromatography using a 75 μ × 150 mm, 5 μ particle porous graphitic carbon (PGC) chip with an integrated 9 mm, 160 nL trapping column. Nucleosides were separated using a formic acid/acetonitrile gradient. The method was able to separate isobaric nucleosides as well as nucleosides with isotopic overlap to allow unambiguous MSn identification on a low resolution ion-trap. Synthesis of 5-hydroxycytidine (oh5C) was achieved from 5-hydroxyuracil in a novel three-step enzymatic process. When operated in its native state using formic acid/acetonitrile, PGC oxidized oh5C to its corresponding glycols and formic acid conjugates. Reduction of the PGC stationary phase was achieved by flushing the chip with 2.5 mM oxalic acid and adding 1 mM oxalic acid to the online solvents. Analyzed under reduced chromatographic conditions oh5C was readily identified by its MH+ m/z 260 and MSn fragmentation pattern. This investigation is, to our knowledge, the first instance where oxalic acid has been used as an online reducing agent for LC/MS. The method was subsequently used for complete characterization of nucleosides found in tRNAs using both PGC and C18 chips.
The pH Dependence of Hairpin Ribozyme Catalysis Reflects Ionization of an Active Site Adenine
J. Biol. Chem. 2011, 286, 17658-17664.
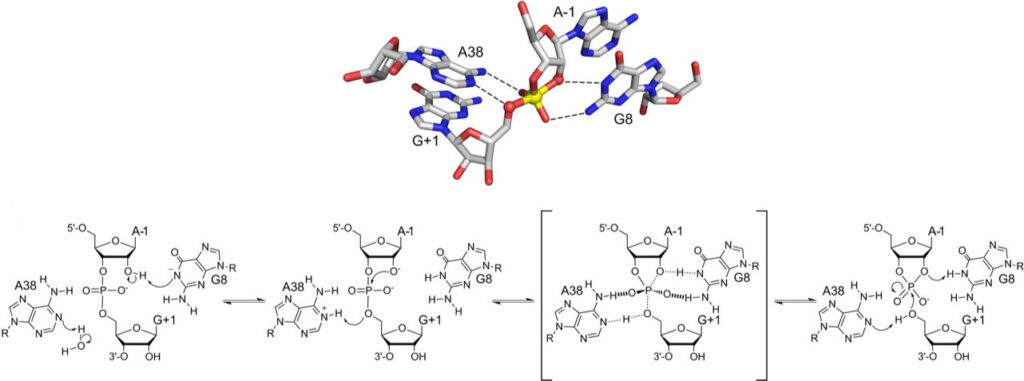
Abstract: Understanding how self-cleaving ribozymes mediate catalysis is crucial in light of compelling evidence that human and bacterial gene expression can be regulated through RNA self-cleavage. The hairpin ribozyme catalyzes reversible phosphodiester bond cleavage through a mechanism that does not require divalent metal cations. Previous structural and biochemical evidence implicated the amidine group of an active site adenosine, A38, in a pH-dependent step in catalysis. We developed a way to determine microscopic pKa values in active ribozymes based on the pH-dependent fluorescence of 8-azaadenosine (8azaA). We compared the microscopic pKa for ionization of 8azaA at position 38 with the apparent pKa for the self-cleavage reaction in a fully functional hairpin ribozyme with a unique 8azaA at position 38. Microscopic and apparent pKa values were virtually the same, evidence that A38 protonation accounts for the decrease in catalytic activity with decreasing pH. These results implicate the neutral unprotonated form of A38 in a transition state that involves formation of the 5′-oxygen-phosphorus bond.
Enzymatic De Novo Pyrimidine Nucleotide Synthesis
J. Am. Chem. Soc. 2011, 133, 297-304.
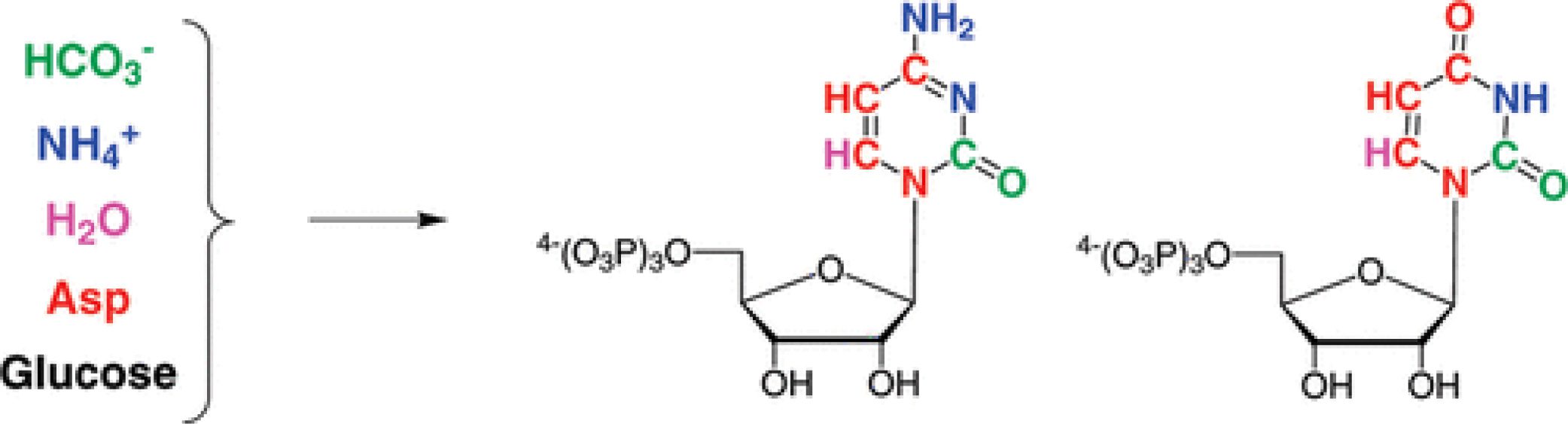
Abstract: The use of stable isotope labeling has revolutionized NMR studies of nucleic acids, and there is a need for methods of incorporation of specific isotope labels to facilitate specific NMR experiments and applications. Enzymatic synthesis offers an efficient and flexible means to synthesize nucleoside triphosphates from a variety of commercially available specifically labeled precursors, permitting isotope labeling of RNAs prepared by in vitro transcription. Here, we recapitulate de novo pyrimidine biosynthesis in vitro, using recombinantly expressed enzymes to perform efficient single-pot syntheses of UTP and CTP that bear a variety of stable isotope labeling patterns. Filtered NMR experiments on 13C,15N,2H-labeled HIV-2 TAR RNA demonstrate the utility and value of this approach. This flexible enzymatic synthesis will make implementing detailed and informative RNA stable isotope labeling schemes substantially more cost-effective and efficient, providing advanced tools for the study of structure and dynamics of RNA molecules.
Active Site Purines and Catalysis of RNA Self‐Cleavage
FASEB J. 2010, 24, 412.
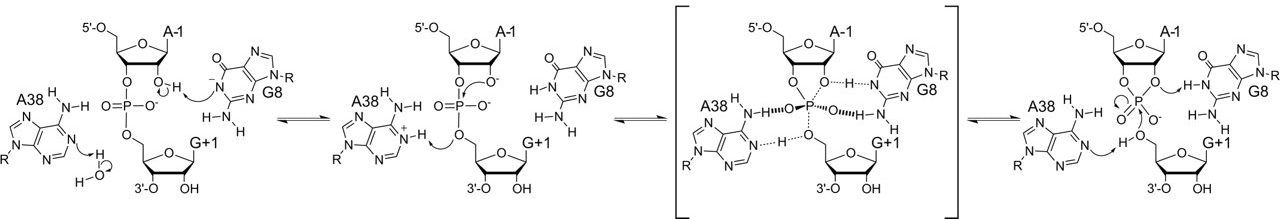
Abstract: The hairpin ribozyme mediates catalysis through nucleotide functional groups, without metal cation cofactors. The positions of G8 and A38 in the ribozyme active site resemble the orientation of two histidines in the active site of ribonuclease A, leading to the model that G8 and A38 mediate catalysis through a similar general acid base mechanism. However, adenosine and guanosine undergo ionization only at pH extremes, at least in solution, which seems to make them poor acid base catalysts. We used 8-azapurine fluorescence to learn whether purine ionization equilibria change in the active site relative to pKa values in solution. 8-azapurines display high fluorescence emission intensity when N1 is deprotonated and low intensity when N1 is protonated. A ribozyme with 8azaG8 exhibits full catalytic activity and cleaves with an apparent pKa value in the neutral range, similar to an unmodified ribozyme. Microscopic pKa values for deprotonation of 8azaG in the active site were about 3 units higher than apparent pKa values determined from the pH dependence of self-cleavage kinetics. Thus, the increase in self-cleavage activity with increasing pH does not reflect G8 deprotonation and G8 is mostly protonated at neutral pH. A simple interpretation of these results is that G8 functions in the protonated form, perhaps by donating hydrogen bonds that stabilize the transition state.
Enzymatic Synthesis and Structural Characterization of C-13, N-15-poly(ADP-Ribose)
J. Am. Chem. Soc. 2009, 40, 14571-14578.
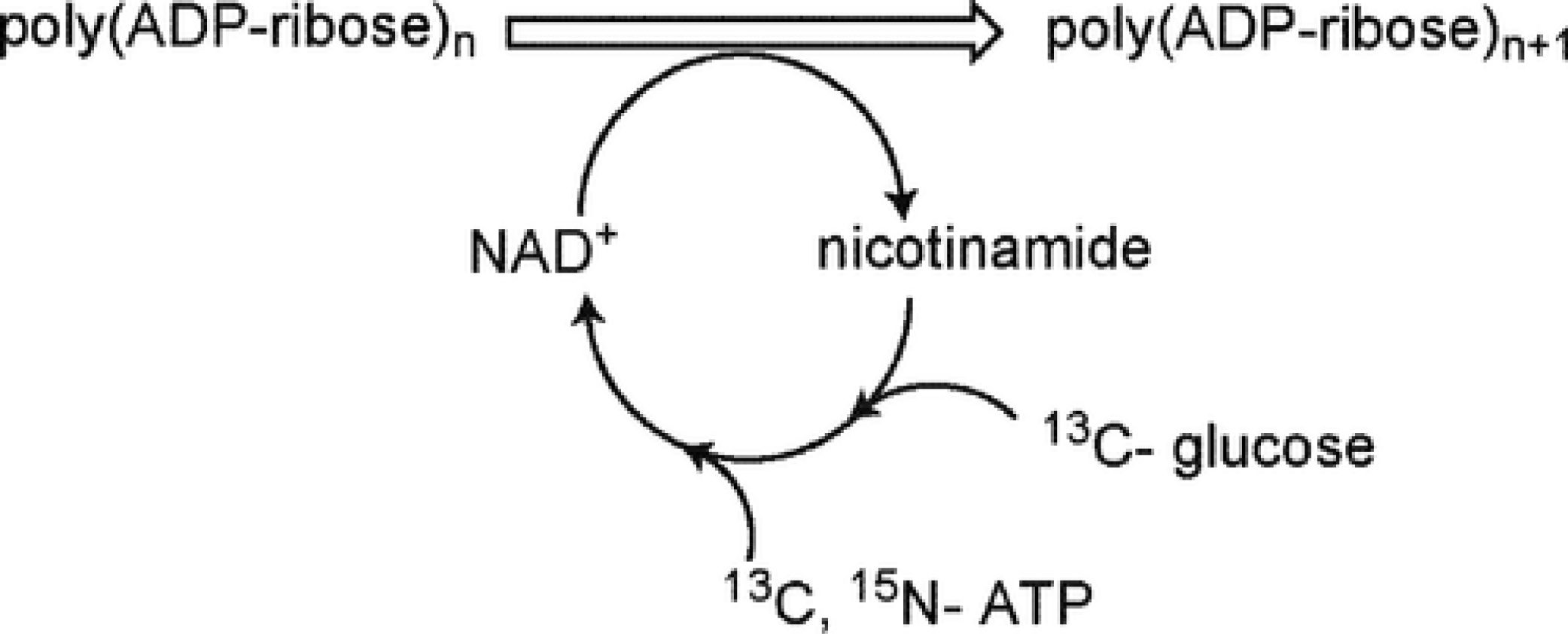
Abstract: Poly(ADP-ribose) is a significant nucleic acid polymer involved with diverse functions in eukaryotic cells, yet no structural information is available. A method for the synthesis of 13C,15N-poly(ADP-ribose) (PAR) has been developed to allow characterization of the polymer using multidimensional nuclear magnetic resonance (NMR) spectroscopy. Successful integration of pentose phosphate, nicotinamide adenine dinucleotide biosynthesis, and cofactor recycling pathways with poly(ADP-ribose) polymerase-1 permitted labeling of PAR from 13C-glucose and 13C,15N-ATP in a single pot reaction. The scheme is efficient, yielding ~400 nmoles of purified PAR from 5 μmoles ATP, and the behavior of the synthetic PAR is similar to data from PAR synthesized by cell extracts. The resonances for 13C,15N-PAR were unambiguously assigned, but the polymer appears to be devoid of inherent regular structure. PAR may form an ordered macromolecular structure when interacting with proteins, and due to the extensive involvement of PAR in cell function and disease, further studies of PAR structure will be required. The labeled PAR synthesis reported here will provide an essential tool for the future study of PAR-protein complexes.
Stable Isotope Pulse-Chase Monitored by Quantitative Mass Spectrometry Applied to E. coli 30S Ribosome Assembly Kinetics
Methods 2009, 49, 136-141.
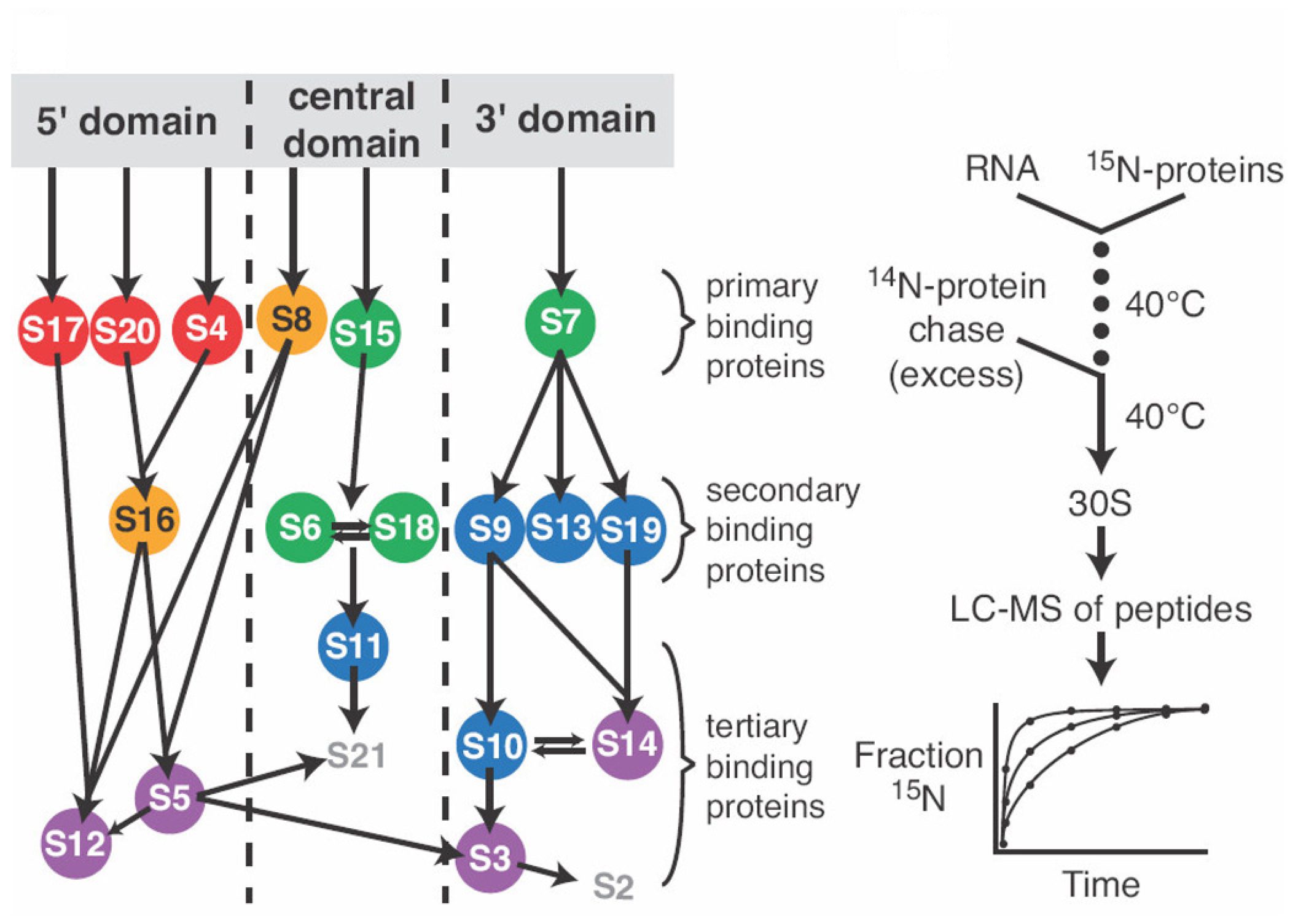
Abstract: Stable isotope mass spectrometry has become a widespread tool in quantitative biology. Pulse-chase monitored by quantitative mass spectrometry (PC/QMS) is a recently developed stable isotope approach that provides a powerful means of studying the in vitro self-assembly kinetics of macromolecular complexes. This method has been applied to the Escherichia coli 30S ribosomal subunit, but could be applied to any stable self-assembling complex that can be reconstituted from its component parts and purified from a mixture of components and complex. The binding rates of 18 out of the 20 ribosomal proteins have been measured at several temperatures using PC/QMS. Here, PC/QMS experiments on 30S ribosomal subunit assembly are described, and the potential application of the method to other complexes is discussed. A variation on the PC/QMS experiment is introduced that enables measurement of kinetic cooperativity between proteins. In addition, several related approaches to stable isotope labeling and quantitative mass spectrometry data analysis are compared and contrasted.
Direct Measurement of the Ionization State of an Essential Guanine in the Hairpin Ribozyme
Nat. Chem. Biol. 2009, 5, 351-357.
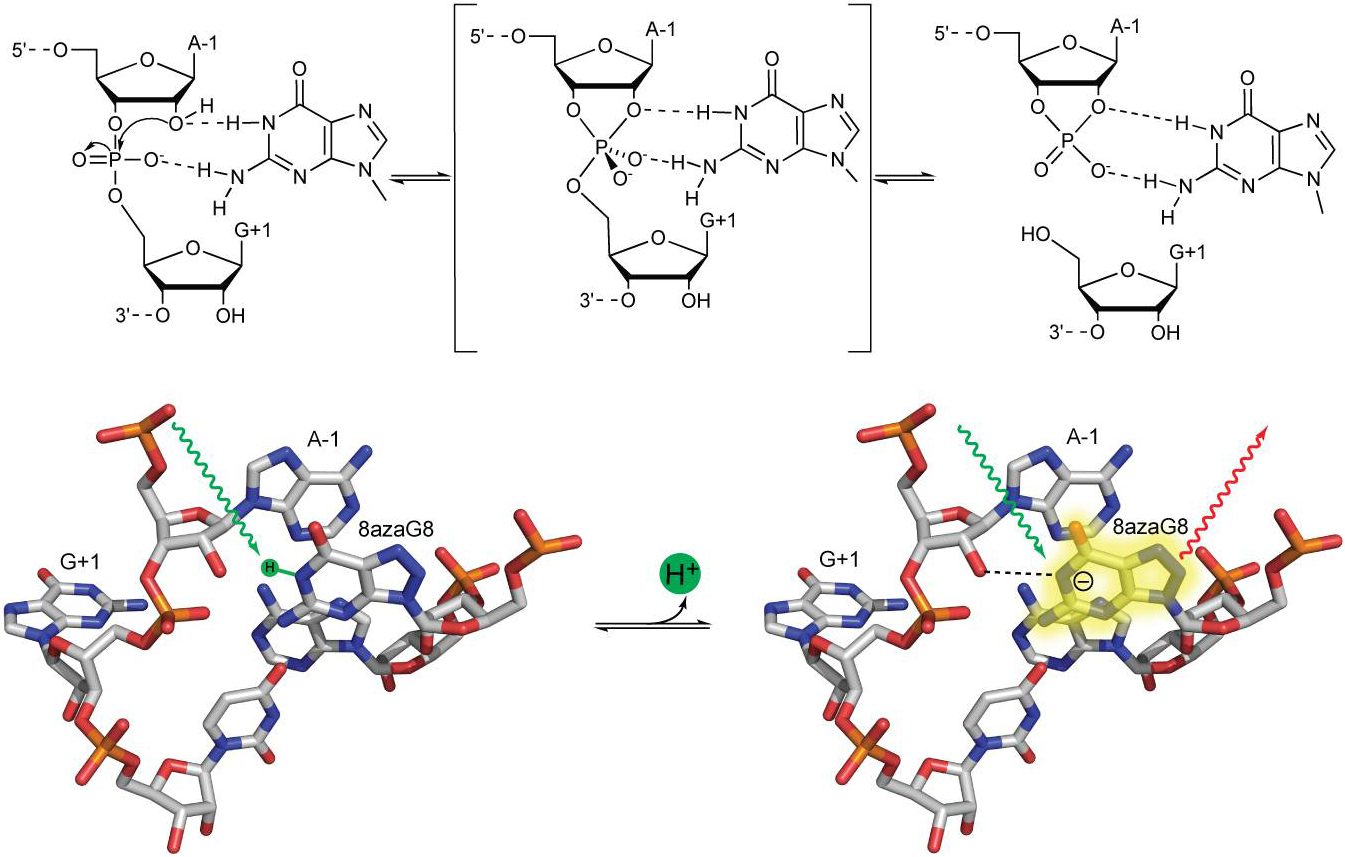
Abstract: Active site guanines are critical for self-cleavage reactions of several ribozymes, but their precise functions in catalysis are unclear. To learn whether protonated or deprotonated forms of guanine predominate in the active site, microscopic pKa values were determined for ionization of 8-azaguanosine substituted for G8 in the active site of a fully functional hairpin ribozyme in order to determine microscopic pKa values for 8-azaguanine deprotonation from the pH dependence of fluorescence. Microscopic pKa values above 9 for deprotonation of 8-azaguanine in the active site were about 3 units higher than apparent pKa values determined from the pH dependence of self-cleavage kinetics. Thus, the increase in activity with increasing pH does not correlate with deprotonation of G8, and most of G8 is protonated at neutral pH. These results do not exclude a role in proton transfer, but a simple interpretation is that G8 functions in the protonated form, perhaps by donating hydrogen bonds.
Pathway Engineered Enzymatic de Novo Purine Nucleotide Synthesis
ACS Chem. Biol. 2008, 3, 499-511.
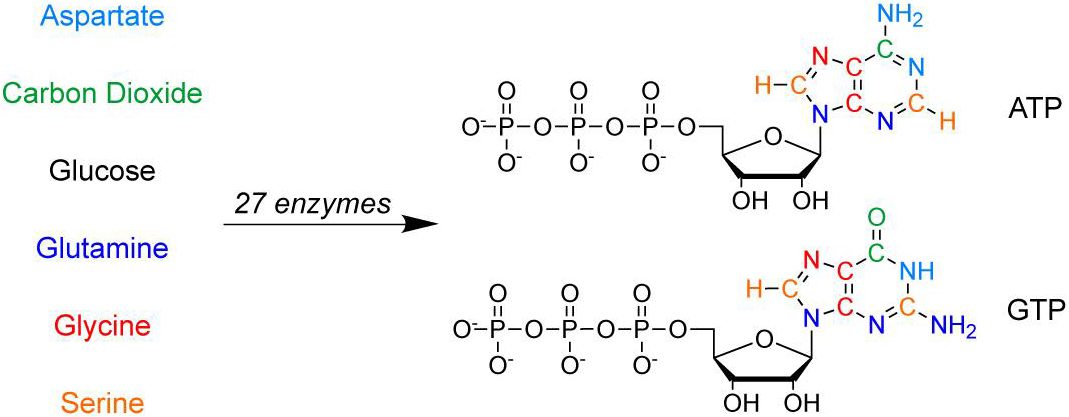
Abstract: A general method for isotopic labeling of the purine base moiety of nucleotides and RNA has been developed through biochemical pathway engineering in vitro. A synthetic scheme was designed and implemented utilizing recombinant enzymes from the pentose phosphate and de novo purine synthesis pathways, with regeneration of folate, aspartate, glutamine, ATP, and NADPH cofactors, in a single-pot reaction. Syntheses proceeded quickly and efficiently in comparison to chemical methods with isolated yields up to 66% for 13C,15N-enriched ATP and GTP. The scheme is robust and flexible, requiring only serine, NH4+, glucose, and CO2 as stoichiometric precursors in labeled form. Using this approach, U-13C-GTP, U-13C,15N-GTP, 13C-2,8-ATP, and U-15N-GTP were synthesized on a millimole scale, and the utility of the isotope labeling is illustrated in NMR spectra of HIV-2 transactivation region RNA containing 13C-2,8-adenosine and 15N-1,N2,3,7,9-guanosine. Pathway engineering in vitro permits complex synthetic cascades to be effected, expanding the applicability of enzymatic synthesis.
Quantitative Analysis of Isotope Distributions in Proteomic Mass Spectrometry Using Least-Squares Fourier Transform Convolution
Analytical Chem. 2008, 80, 4906-4917.
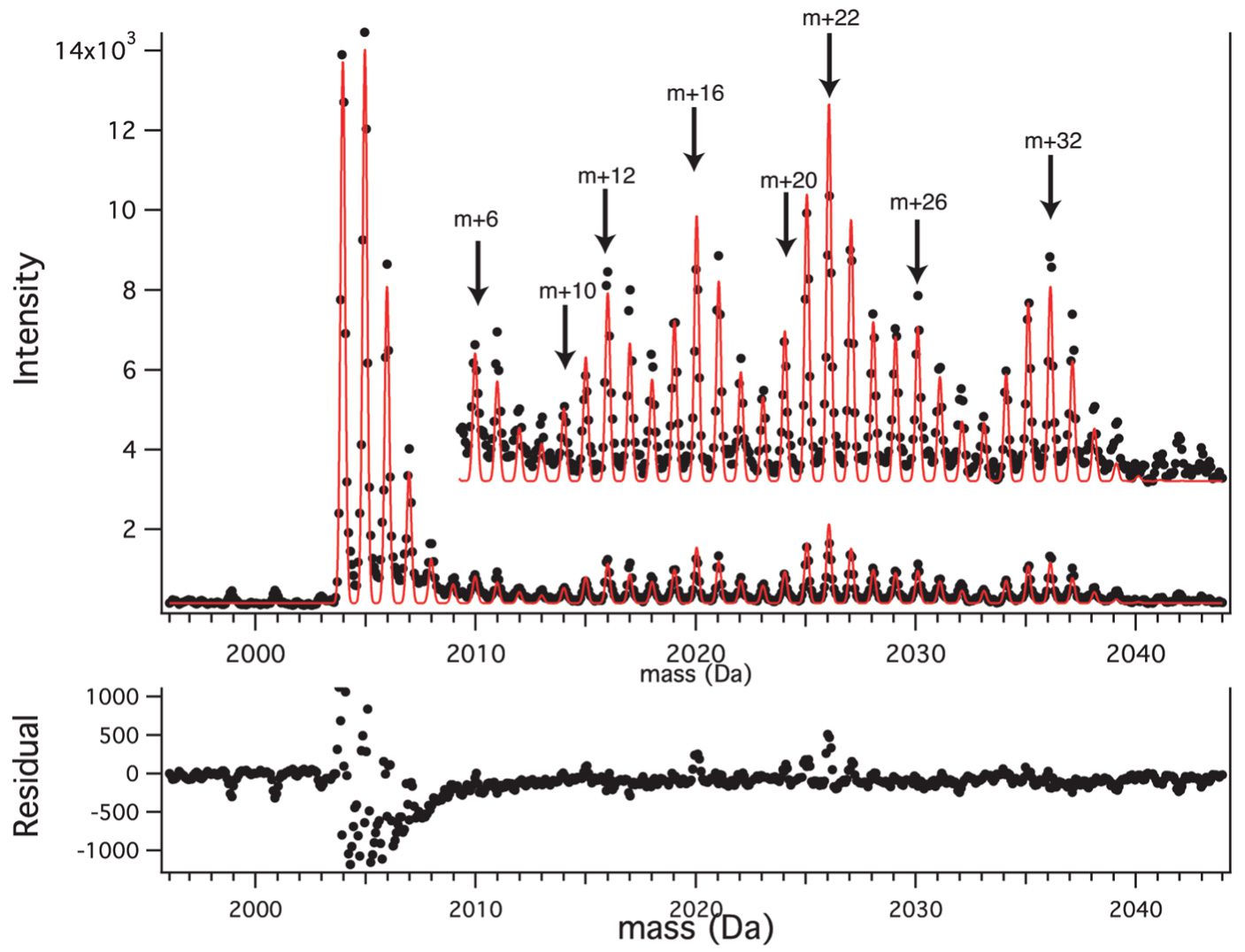
Abstract: Quantitative proteomic mass spectrometry involves comparison of the amplitudes of peaks resulting from different isotope labeling patterns, including fractional atomic labeling and fractional residue labeling. We have developed a general and flexible analytical treatment of the complex isotope distributions that arise in these experiments, using Fourier transform convolution to calculate labeled isotope distributions and least-squares for quantitative comparison with experimental peaks. The degree of fractional atomic and fractional residue labeling can be determined from experimental peaks at the same time as the integrated intensity of all of the isotopomers in the isotope distribution. The approach is illustrated using data with fractional (15)N-labeling and fractional (13)C-isoleucine labeling. The least-squares Fourier transform convolution approach can be applied to many types of quantitative proteomic data, including data from stable isotope labeling by amino acids in cell culture and pulse labeling experiments.
Envelope: Interactive Software for Modeling and Fitting Complex Isotope Distributions
BMC Bioinformatics 2008, 9, 446-455.
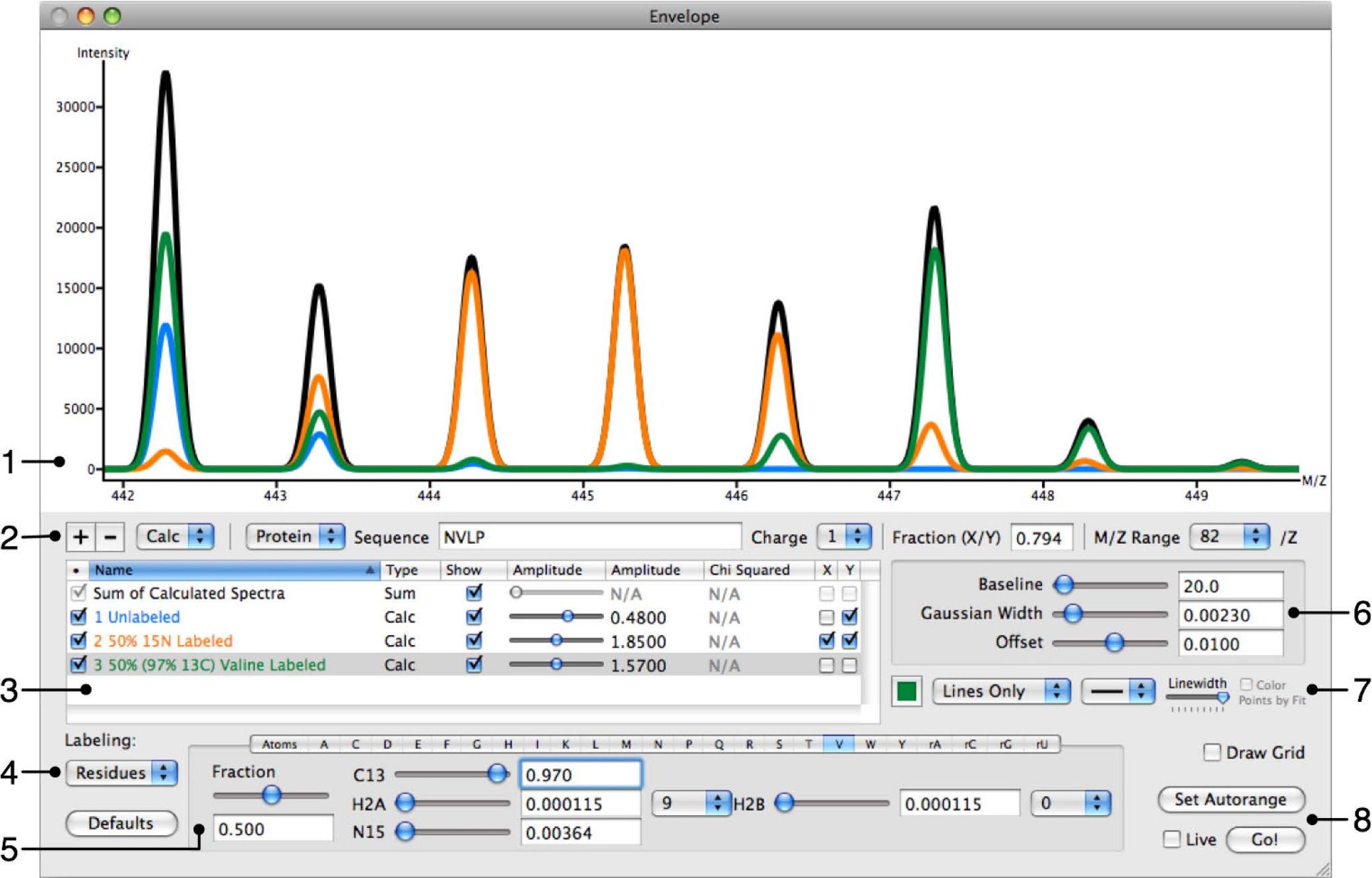
Abstract: An important aspect of proteomic mass spectrometry involves quantifying and interpreting the isotope distributions arising from mixtures of macromolecules with different isotope labeling patterns. These patterns can be quite complex, in particular with in vivo metabolic labeling experiments producing fractional atomic labeling or fractional residue labeling of peptides or other macromolecules. In general, it can be difficult to distinguish the contributions of species with different labeling patterns to an experimental spectrum and difficult to calculate a theoretical isotope distribution to fit such data. There is a need for interactive and user-friendly software that can calculate and fit the entire isotope distribution of a complex mixture while comparing these calculations with experimental data and extracting the contributions from the differently labeled species. Envelope has been developed to be user-friendly while still being as flexible and powerful as possible. Envelope can simultaneously calculate the isotope distributions for any number of different labeling patterns for a given peptide or oligonucleotide, while automatically summing these into a single overall isotope distribution. Envelope can handle fractional or complete atom or residue-based labeling, and the contribution from each different user-defined labeling pattern is clearly illustrated in the interactive display and is individually adjustable. At present, Envelope supports labeling with 2H, 13C, and 15N, and supports adjustments for baseline correction, an instrument accuracy offset in the m/z domain, and peak width. Furthermore, Envelope can display experimental data superimposed on calculated isotope distributions, and calculate a least-squares goodness of fit between the two. All of this information is displayed on the screen in a single graphical user interface. Envelope supports high-quality output of experimental and calculated distributions in PNG or PDF format. Beyond simply comparing calculated distributions to experimental data, Envelope is useful for planning or designing metabolic labeling experiments, by visualizing hypothetical isotope distributions in order to evaluate the feasibility of a labeling strategy. Envelope is also useful as a teaching tool, with its real-time display capabilities providing a straightforward way to illustrate the key variable factors that contribute to an observed isotope distribution. Envelope is a powerful tool for the interactive calculation and visualization of complex isotope distributions for comparison to experimental data.
RNA Structure Determination by NMR
Methods Mol. Biol. 2008, 452, 29-61.
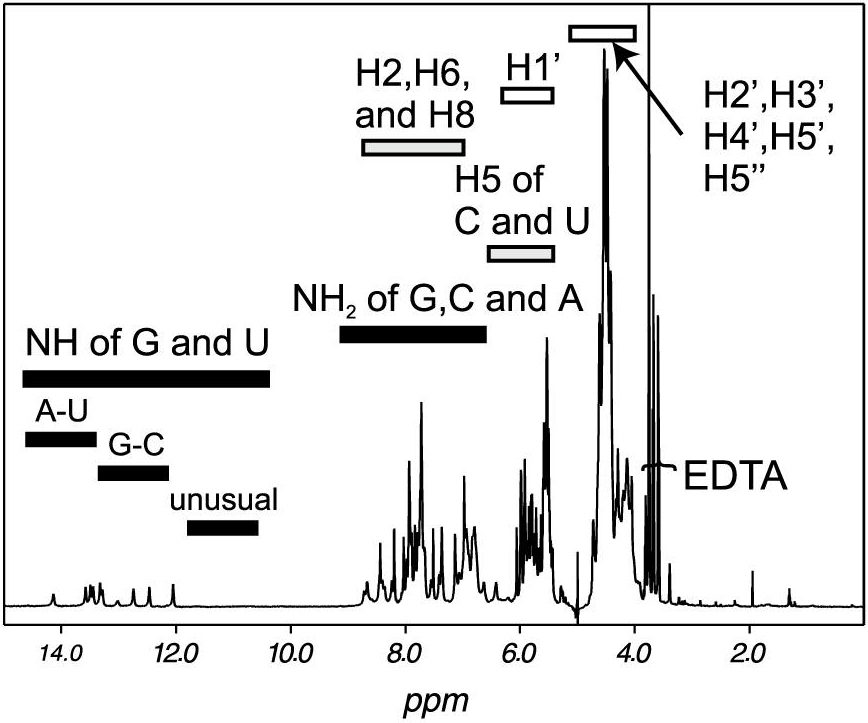
Abstract: This chapter reviews the methodologies for RNA structure determination by liquid-state nuclear magnetic resonance (NMR). The routine production of milligram quantities of isotopically labeled RNA remains critical to the success of NMR-based structure studies. The standard method for the preparation of isotopically labeled RNA for structural studies in solution is in vitro transcription from DNA oligonucleotide templates using T7 RNA polymerase and unlabeled or isotopically labeled nucleotide triphosphates (NTPs). The purification of the desired RNA can be performed by either denaturing polyacrylamide gel electrophoresis (PAGE) or anion-exchange chromatography. Our basic strategy for studying RNA in solution by NMR is outlined. The topics covered include RNA resonance assignment, restraint collection, and the structure calculation process. Selected examples of NMR spectra are given for a correctly folded 30 nucleotide-containing RNA.
Synthesis of 5-Fluoropyrimidine Nucleotides as Sensitive NMR Probes of RNA Structure
J. Am. Chem. Soc. 2007, 129, 14911-14921.
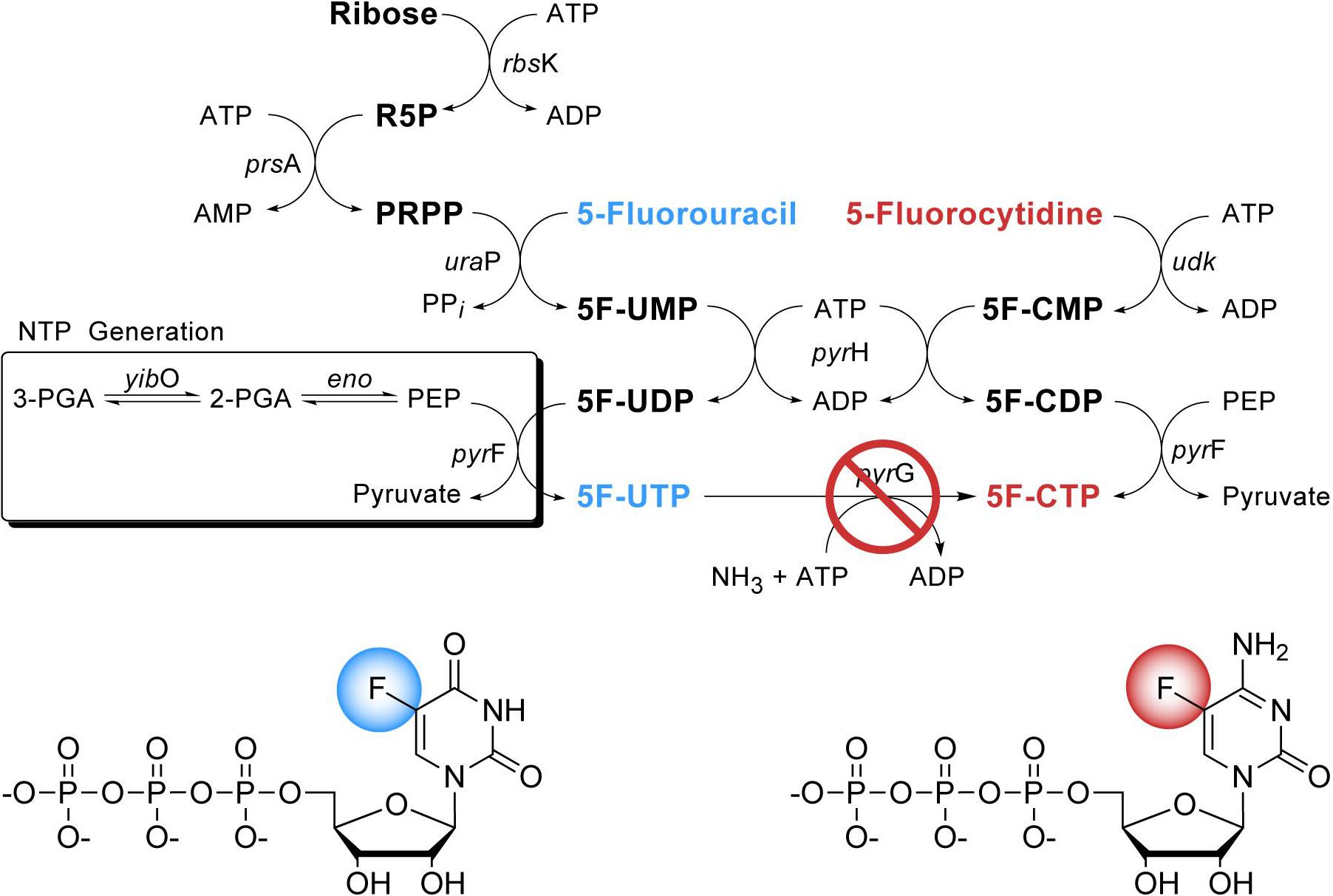
Abstract: Enzymatic synthesis methods for the fluorinated 5′-triphosphate analogues 5F-UTP and 5F-CTP have been developed to facilitate 19F-labeling of RNAs for biophysical studies. HIV-2 TAR RNAs were synthesized using these analogues by in vitro transcription reactions using T7 RNA polymerase. The uniform incorporation of 5F-U or 5F-C analogues into HIV-2 TAR RNA transcripts does not significantly alter the RNA structure or thermodynamic stability. Fluorine observed homonuclear 19F-19F and heteronuclear 19F−1H NOE experiments providing selective distance information are presented and discussed. The availability of efficient synthesis of 5F-UTP, and for the first time, 5F-CTP, will facilitate the use of 5F-labeled RNAs in structural, ligand binding, and dynamic studies of RNAs using the advantages of 19F-labeling.
Strong Coupling Effects During X-pulse CPMG Experiments Recorded on Heteronuclear ABX Spin Systems: Artifacts and a Simple Solution
J. Biomol. NMR 2007, 38, 41-46.
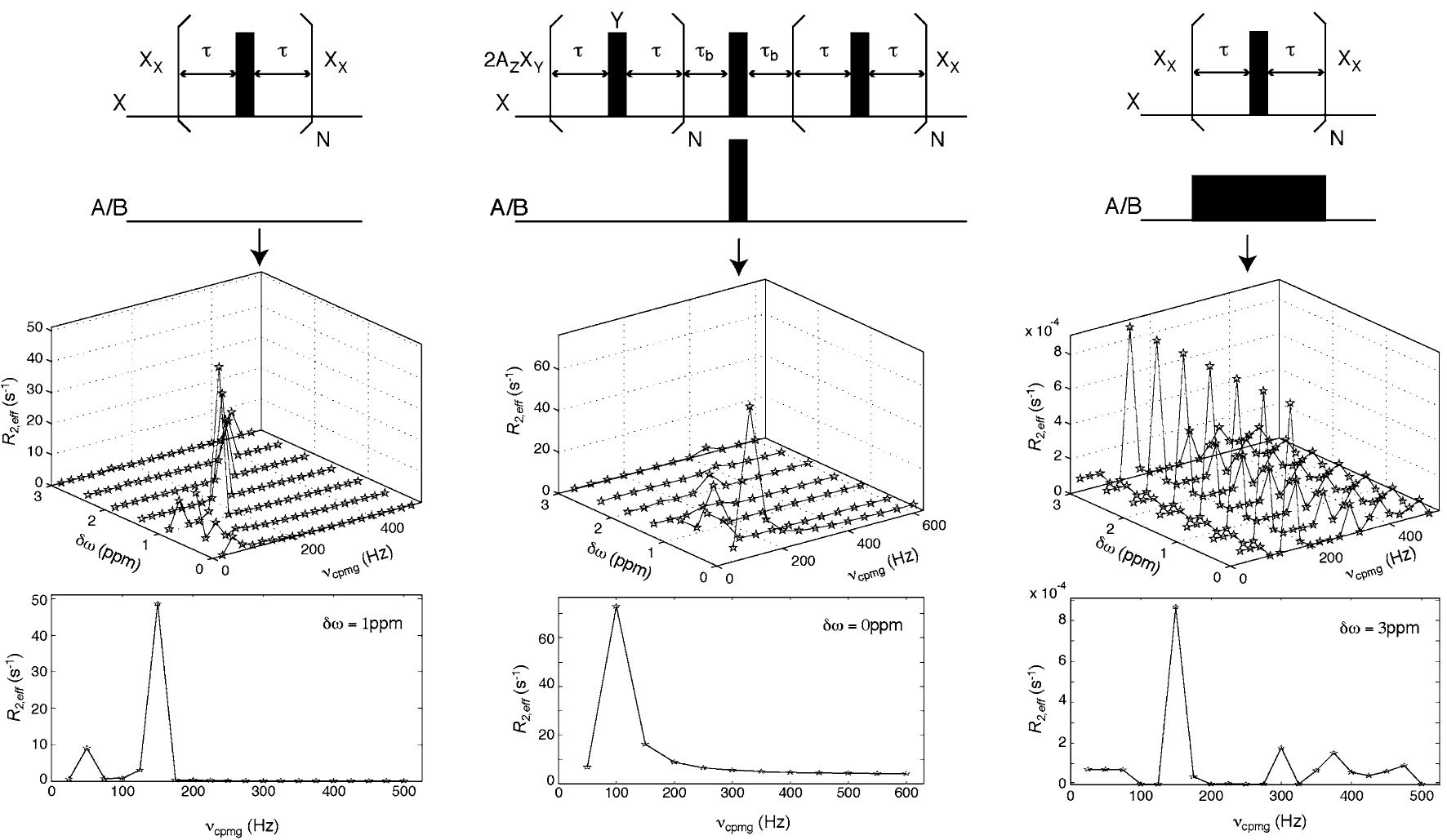
Abstract: Simulation and experiment have been used to establish that significant artifacts can be generated in X-pulse CPMG relaxation dispersion experiments recorded on heteronuclear ABX spin-systems, such as 13Ci–13Cj–1H, where 13Ci and 13Cj are strongly coupled. A qualitative explanation of the origin of these artifacts is presented along with a simple method to significantly reduce them. An application to the measurement of 1H CPMG relaxation dispersion profiles in an HIV-2 TAR RNA molecule where all ribose sugars are protonated at the 2′ position, deuterated at all other sugar positions and 13C labeled at all sugar carbons is presented to illustrate the problems that strong 13C-13C coupling introduces and a simple solution is proposed.
8-Azaguanine Reporter of Purine Ionization States in Structured RNAs
J. Am. Chem. Soc. 2007, 129, 3426-3432.
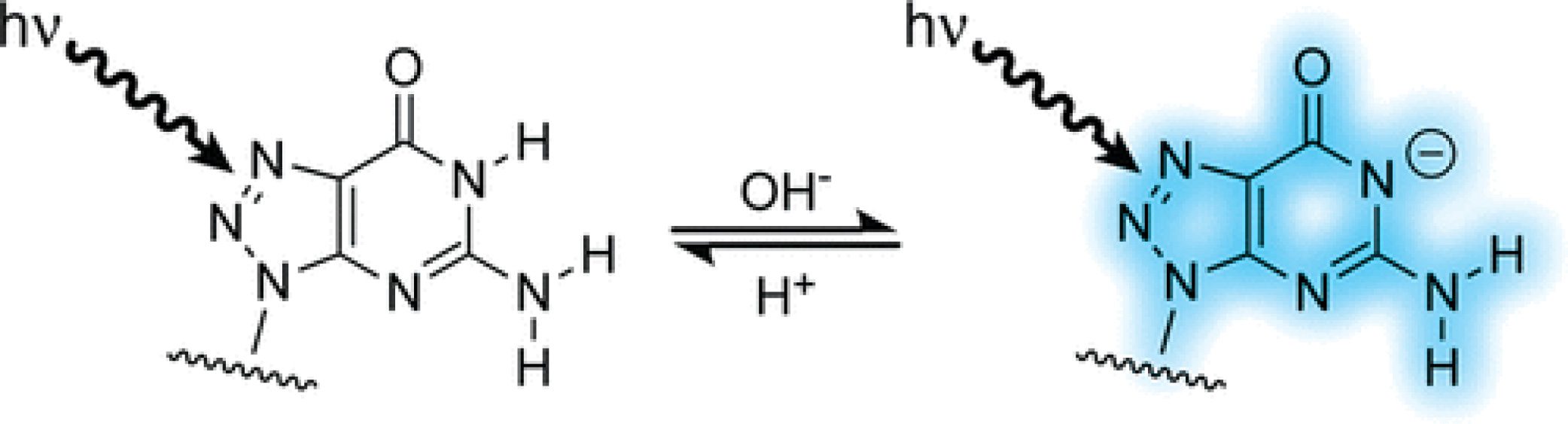
Abstract: The fluorescent nucleotide analogue 8-azaguanosine-5‘-triphosphate (8azaGTP) is prepared easily by in vitro enzymatic synthesis methods. 8azaGTP is an efficient substrate for T7 RNA polymerase and is incorporated specifically opposite cytosine in the transcription template, as expected for a nucleobase analogue with the same Watson−Crick hydrogen bonding face as guanine. 8-Azaguanine (8azaG) in oligonucleotides also is recognized as guanine during ribonuclease T1 digestion. Moreover, replacement of guanine by 8azaG does not alter the melting temperature of base-paired RNAs significantly, evidence that 8azaG does not disrupt stacking and hydrogen bonding interactions. 8azaGTP displays a high fluorescent quantum yield when the N1 position is deprotonated at high pH, but fluorescence intensity decreases significantly when N1 is protonated at neutral pH. Fluorescence is quenched 10-fold to 100-fold when 8azaG is incorporated into base-paired RNA and remains pH-dependent, although apparent pKa values determined from the pH dependence of fluorescence intensity shift in the basic direction. Thus, 8azaG is a guanine analogue that does not perturb RNA structure and displays pH-dependent fluorescence that can be used to probe the ionization states of nucleobases in structured RNAs. A key application will be in determining the ionization state of active site nucleobases that have been implicated in the catalytic mechanisms of RNA enzymes.
New RNA Labeling Methods Offer Dramatic Sensitivity Enhancements in 2H NMR Relaxation Spectra
J. Am. Chem. Soc. 2006, 128, 9346-9347.
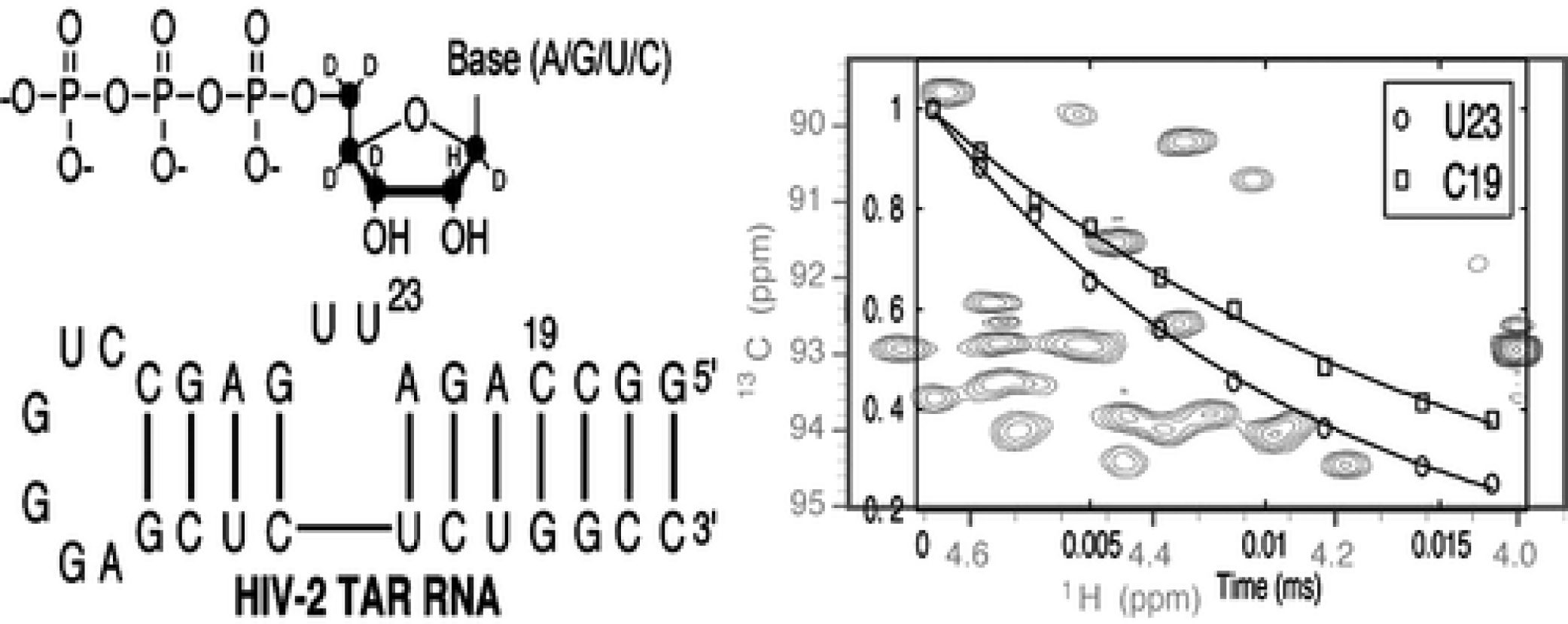
Abstract: A new labeling strategy is presented that greatly facilitates the measurement of 2H spin relaxation rates in RNA molecules as a probe of pico- to nanosecond time scale dynamics. In this labeling scheme the sugar positions are uniformly 13C-labeled, with position 2′ protonated and all other sites on the sugar deuterated. Pulse sequences are presented for measurement of 2H R1 and R2 relaxation rates at positions 1′, 3′, and 4′ with sensitivity gains that are on the order of 5-fold relative to previous methods that employed random fractional deuteration. The improved sensitivity is transformative and facilitates the study of motion in moderately sized RNA molecules with good sensitivity. The utility of the approach is demonstrated with an application to HIV-2 TAR, where the site-specific measures of molecular dynamics at sugar positions obtained here complement previous studies of dynamics at aromatic sites in the molecule.
Measurement of Long-Range 1H-19F Scalar Coupling Constants and Their Glycosidic Torsion Dependence in 5-Fluoropyrimidine-Substituted RNA
J. Am. Chem. Soc. 2006, 128, 5851-5858.
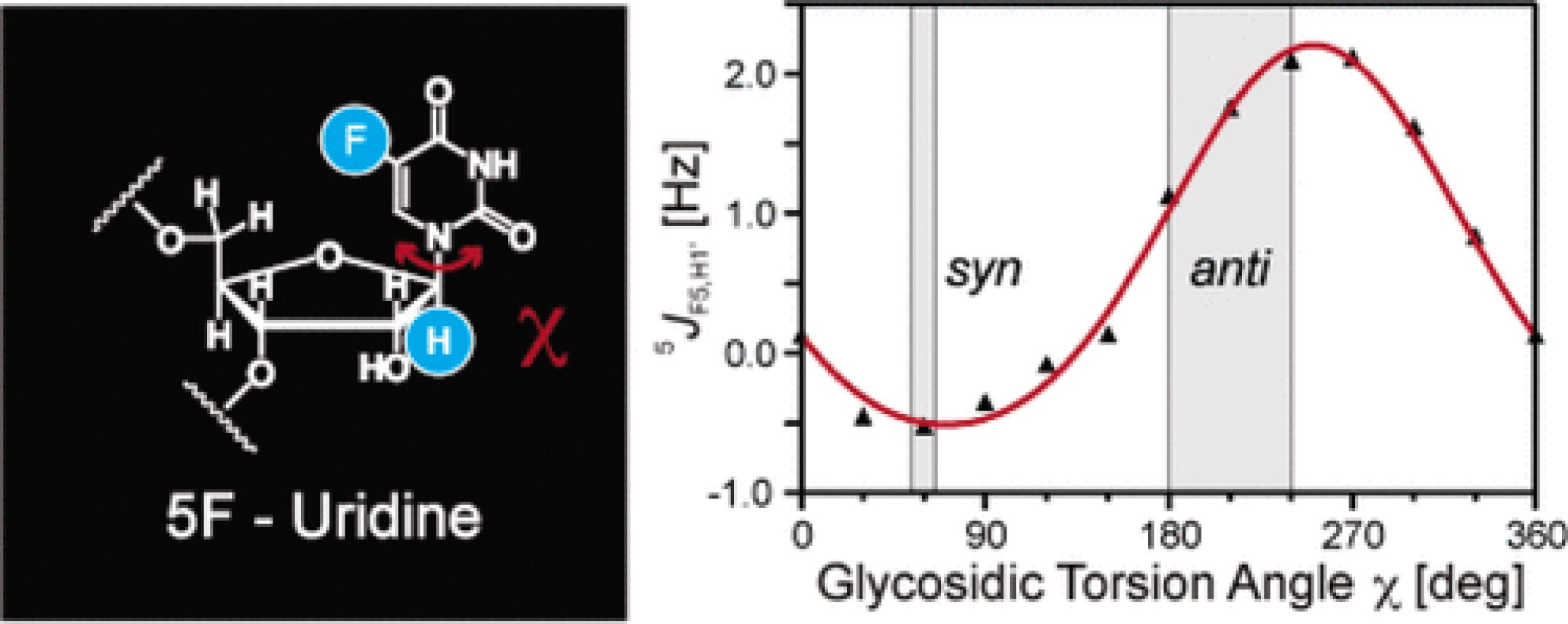
Abstract: Long-range scalar 5J(H1′,F) couplings were observed in 5-fluoropyrimidine-substituted RNA. We developed a novel S3E-19F-α,β-edited NOESY experiment for quantitation of these long-range scalar 5J(H1′,F) couplings, where the J-couplings can be extracted from inspection of intraresidual (H1′,H6) NOE cross-peaks. Quantum chemical calculations were exploited to investigate the relation between scalar couplings and conformations around the glycosidic bond in oligonucleotides. The theoretical dependence of the observed 5J(H1′,F) couplings on the torsion angle χ can be described by a generalized Karplus relationship. The corresponding density functional theory (DFT) analysis is outlined. Additional NMR experiments facilitating the resonance assignments of 5-fluoropyrimidine-substituted RNAs are described, and chemical shift changes due to altered shielding in the presence of fluorine-19 (19F) are presentes.
RNA Helical Packing in Solution: NMR Structure of a 30 kDa GAAA Tetraloop-Receptor Complex
J. Mol. Biol. 2005, 351, 371-382.
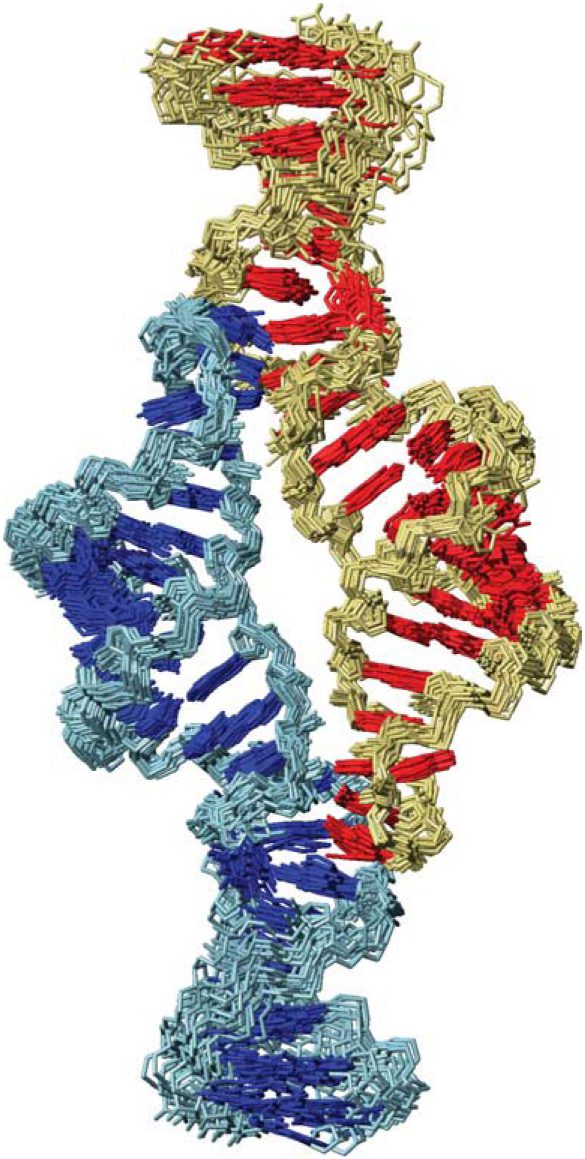
Abstract: Tertiary interactions are critical for proper RNA folding and ribozyme catalysis. RNA tertiary structure is often condensed through long-range helical packing interactions mediated by loop-receptor motifs. RNA structures displaying helical packing by loop-receptor interactions have been solved by X-ray crystallography, but not by NMR. Here, we report the NMR structure of a 30 kDa GAAA tetraloop-receptor RNA complex. In order to stabilize the complex, we used a modular design in which the RNA was engineered to form a homodimer, with each subunit containing a GAAA tetraloop phased one helical turn apart from its cognate 11-nucleotide receptor domain. The structure determination utilized specific isotopic labeling patterns (2H, 13C, and 15N) and refinement against residual dipolar couplings. We observe a unique and highly unusual chemical shift pattern for an adenosine platform interaction that reveals a spectroscopic fingerprint for this motif. The structure of the GAAA tetraloop-receptor interaction is well defined solely from experimental NMR data, shows minor deviations from previously solved crystal structures, and verifies the previously inferred hydrogen bonding patterns within this motif. This work demonstrates the feasibility of using engineered homodimers as modular systems for the determination of RNA tertiary interactions by NMR.
Enzymatic Synthesis and 19F NMR Studies of 2-Fluoroadenine-Substituted RNA
J. Am. Chem. Soc. 2004, 126, 11776-11777.
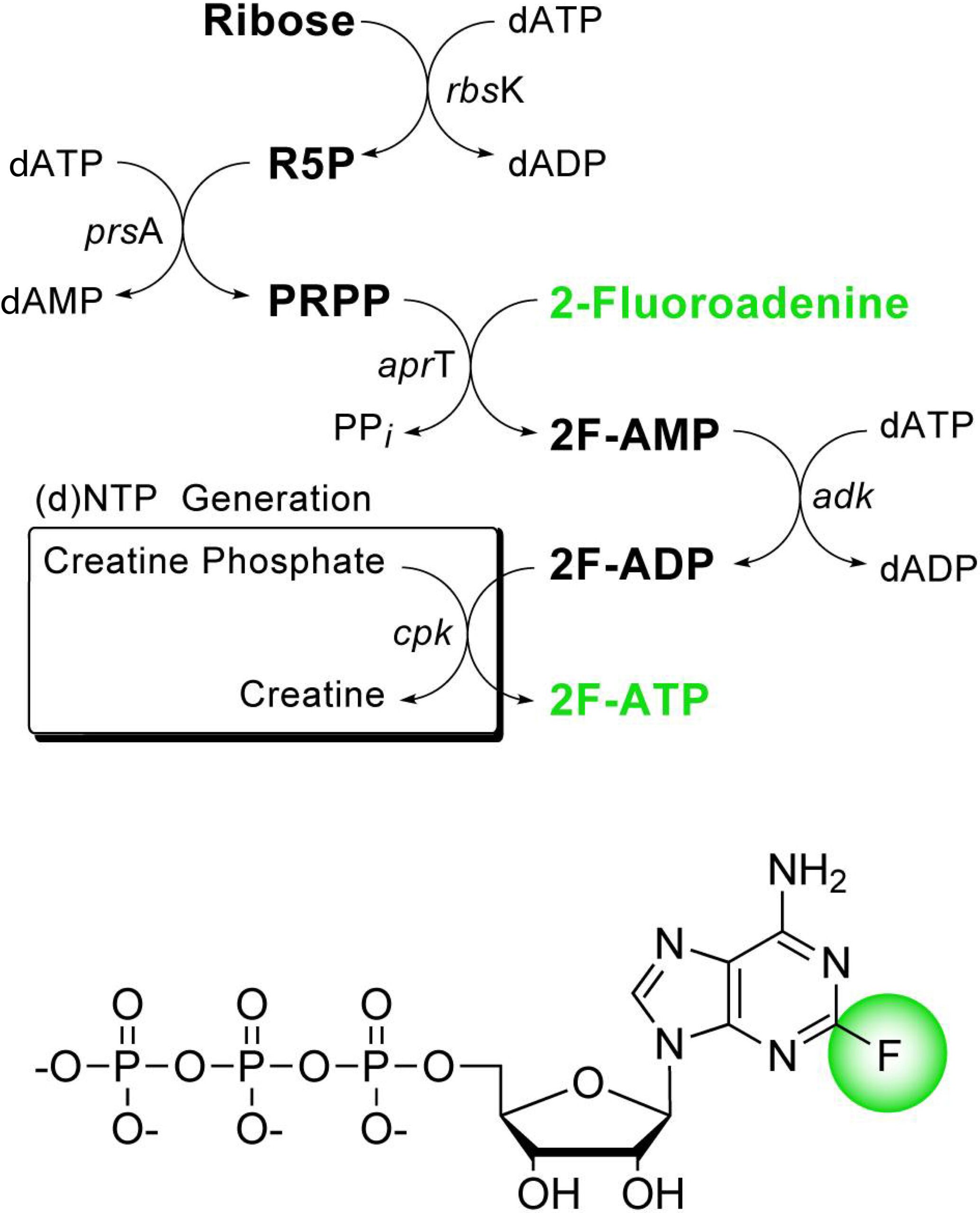
Abstract: The production of isotopically labeled RNA remains critical to current NMR structural studies. One approach to obtain simple NMR spectra is to label with a nucleus that is not naturally occurring in RNA. Fluorine-19 can serve as a sensitive site-specific probe upon incorporation into RNA. Here we report the efficient in vitro enzymatic synthesis of 2-fluoroadenosine-5′-triphosphate and its incorporation into the HIV-2 transactivation region (TAR) of RNA by DNA template-directed transcription using phage T7 RNA polymerase. We provide unequivocal evidence for this 19F-substituted base analogue capability to selectively interact with uracil, forming 2F-A−U base pairs in RNA. The introduction of a 2-fluoroadenyl substitution is relatively nonperturbing and provides us with uniquely positioned, sensitive NMR reporter groups to monitor structural changes in the local RNA environment.
Improvement in the Apparent Mass Resolution of Oligonucleotides by Using 12C/14N-Enriched Samples
Analytical Chem. 2002, 74, 226-231.
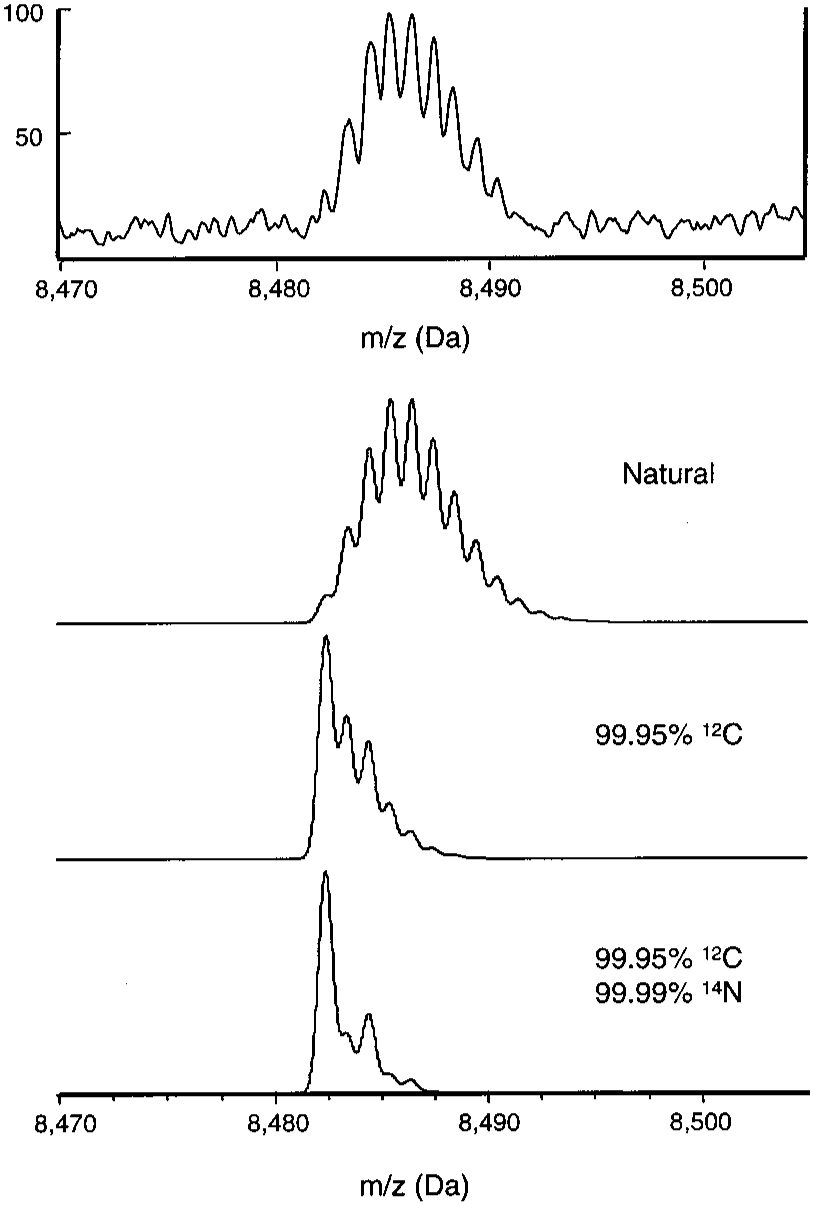
Abstract: The apparent mass resolution of oligonucleotides in time-of-flight (TOF) mass spectrometers has been examined. In a reflectron TOF instrument, where the isotopic profile can be completely resolved, the apparent resolution matches the instrument’s resolving power. In a linear TOF instrument, unresolved isotopic profiles limit the apparent resolution to much lower values than the actual instrument resolution. By using 12C/14N-enriched oligonucleotides, the apparent resolution can be improved significantly. The isotope enrichment method also enhances the signal-to-noise ratio.
Preparation of Specifically 2H- and 13C-Labeled Ribonucleotides
Methods Enzymol. 2000, 317, 18-38.
Abstract: This chapter discusses the enzymatic synthesis of specifically labeled ribonucleotide triphosphates (NTPs) for the in vitro transcription of RNA strategy. Isotopically labeled RNAs are prepared in transcription reactions using labeled NTPs and T7 RNA polymerase. Uniformly labeled NTPs are readily produced by the phosphorylation of nucleotides isolated from bacterial cultures. While some specific isotopic labeling patterns have been produced by bacterial growth on specifically labeled substrates, a general biochemical synthesis of labeled NTPs offers the advantage of a diversity of isotopic labeling patterns that can be created without metabolic scrambling. By coupling the enzymes from the glycolysis and pentose phosphate pathways with those of nucleotide biosynthesis and salvage, isotopically labeled glucose can be converted into NTPs. The strategy for enzymatic synthesis involves the conversion of glucose into 5-phospho-D-ribosyl-α-1-pyrophosphate (PRPP), which is then converted to three of the nucleoside triphosphates—adenosine triphosphate (ATP), guanosine triphosphate (GTP), and uracil triphosphate (UTP). Cytosine triphosphate (CTP) is prepared from UTP in a second enzymatic reaction catalyzed by CTP synthase (PYRG).
3D C(CC)H TOCSY Experiment of Assigning Protons and Carbons in Uniformly 13C and Selective 2H Labeled RNA
J. Magn. Reson. 1998, 130, 97-101.
Abstract: We have prepared RNAs uniformly 13C-labeled in the ribose ring and with the H3′, H4′, H5′/H5′′ protons specifically replaced with deuterons (3′, 4′, 5′/5′′-2H-13C, termed d4–13C ribose), which offers the advantage of spectral simplification without sacrifice of sensitivity for the remaining protons. A 3D C(CC)H TOCSY experiment that uses a combination of cross-polarization transfer and deuterium decoupling improves the transfer of magnetization around the ribose ring and facilitates assignment of all ribose carbons and H1′ and H2′ protons in a 30-nucleotide RNA from HIV-2. These and other combinations of labeling and pulse sequence methodology should prove invaluable for the study of large RNAs, RNA-ligand, and RNA-protein complexes.
Preparation of Specifically Deuterated and 13C-Labeled RNA for NMR Studies Using Enzymatic Synthesis
J. Am. Chem. Soc. 1997, 119, 12100-12108.
Abstract: The enzymatic conversion of glucose into ATP, GTP, UTP, and CTP with several different isotopic labeling patterns is described. Enzymes of the pentose phosphate pathway and enzyme-catalyzed hydrogen exchange were used to convert three types of isotopically labeled glucose into [1′,2′,3′,4′,5′,5′′-2H6]NTPs (1−4), [3′,4′,5′,5′′-2H4]UTP (5), [1′,2′,3′,4′,5′-13C5]NTPs (6−9), and [3′,4′,5′,5′′-2H4-1′,2′,3′,4′,5′-13C5]NTPs (10−13), which were then used to synthesize a 30 nucleotide HIV TAR RNA. Representative NOESY and HSQC spectra were acquired to demonstrate the utility of the new labeling patterns. The spectral editing afforded by 2H and 13C labeling dramatically simplifies the crowded NOESY and HSQC spectra of RNA molecules. The synthetic methods described here will permit the preparation of several specifically deuterated and/or 13C-labeled forms of RNA which should be useful in NMR structural studies of large RNAs.
Preparation of Specifically Deuterated RNA for NMR Studies Using a Combination of Chemical and Enzymatics Synthesis
J. Am. Chem. Soc. 1996, 118, 7929–7940.
Abstract: The syntheses of ATP, GTP, UTP, and CTP with deuterium labels on the 3′, 4′, and 5′ carbons (2−5) is described. A combination of chemical and enzymatic synthesis is used where d,l-ribose-3,4,5′,5′′-d4 (±1) is first produced from glycerol-d8 by chemical methods, and then the four 3′,4′,5′,5′′-labeled NTPs (2–5) are prepared from (-1) using enzymes from the purine salvage and pyrimidine biosynthetic metabolic pathways. New procedures were developed for the large scale preparation of GTP and CTP, and existing procedures were modified for the preparation of ATP and UTP. A 30-nucleotide RNA derived from the HIV-2 TAR RNA was prepared with unlabeled NTPs and deuterated NTPs (2–5) to illustrate the dramatic effects of deuteration on the NMR spectra of RNA. The NOESY spectra of the deuterated RNA exhibits greatly reduced spectral crowding compared to that of the unlabeled RNA, and assignment of NOEs to the H2′ protons is simplified due to the specific deuteration pattern. Also, the nonselective T1 and T2 relaxation rates were measured for the deuterated RNA and found to be approximately twice as long as the T1 and T2 relaxation rates of the unlabeled RNA. The spectral simplification and improved relaxation properties of the deuterated RNA should prove useful in the study of large RNAs by NMR.
Improved Large Scale Culture of Methylophilus methylotrophus for 13C/15N Labeling and Random Fractional Deuteration of Ribonucleotides
Nucleic Acids Res. 1996, 24, 4836–4837.
Abstract: Isotopic labeling of RNA with 13C and 15N has become a routine procedure in structural studies by NMR spectroscopy. The methodology in this paper describes the random fractional deuteration of RNA using the obligate methylotropic bacterium, Methylophilus methylotrophus. This bacterium was grown using a non-deuterated carbon source in 52:48 D2O/H2O and we have shown that all protons in the ribonucleotides except for the ribose H1′ become 52% randomly fractionally deuterated. Improved growth conditions for this organism are also described that yield higher cell densities in liquid culture, which is applicable for all labeling procedures.
Preparation of Isotopically Enriched RNAs for Heteronuclear NMR
Methods Enzymol. 1995, 261, 300-322.
Abstract: This chapter discusses a detailed procedure for the production of 13C- and/or 15N-labeled RNA from inexpensive and readily available sources of these isotopes. The ability to overexpress proteins, isotopically labeled with 13C and 15N, is a cornerstone of the current nuclear magnetic resonance (NMR) methodology used to solve solution structures of large proteins. Recently, similar heteronuclear NMR techniques have been applied to the RNA structures in solution. A number of laboratories have developed techniques for the synthesis of isotopically labeled ribonucleotide triphosphates as precursors for the preparation of any RNA of defined sequence and for the efficient assignment of labeled RNAs. Bacterial cells are grown in a minimal salts medium, containing isotopically labeled carbon and/or nitrogen substrates. E. coli are grown to produce 15N-labeled nucleotides, whereas 13C-labeled or 13C/15N nucleotides are best produced, by growing Methylophilus methylotrophus on 13C-methanol that is an economical source of 13C. The cells are harvested and lysed by detergent, the proteins are removed by phenol-chloroform extraction, and the total nucleic acids are precipitated with isopropanol. The boronate chromatography procedure allows one to quantitatively and reproducibly separate deoxyribonucleotides from ribonucleotides. This chromatography, however, must be performed carefully if one is to achieve these results.
Preparation of Isotopically Labeled Ribonucleotides for Multidimensional NMR Spectroscopy of RNA
Nucleic Acids Res. 1992, 20, 4515-4523.
Abstract: A general method for large scale preparation of uniformly isotopically labeled ribonucleotides and RNAs is described. Bacteria are grown on isotopic growth medium, and their nucleic acids are harvested and degraded to mononucleotides. These are enzymatically converted into ribonucleoside triphosphates, which are used in transcription reactions in vitro to prepare RNAs for NMR studies. For 15N-labeling, E. coli is grown on 15N-ammonium sulfate, whereas for 13C-labeling, Methylophilus methylotrophus is grown on 13C-methanol, which is more economical than 13C-glucose. To demonstrate the feasibility and utility of this method, uniformly 13C-labeled ribonucleotides were used to synthesize a 31 nucleotide HIV TAR RNA that was analyzed by 3D-NMR. This method should find widespread use in the structural analysis of RNA by NMR.
Synthesis of a Thymidine Phosphoramidite Labeled with C-13 at C6 - Relaxation Studies of the Loop Region in a C-13 Labeled DNA Hairpin
Nucleic Acids Res. 1988, 16, 1529-1540.
Abstract: A thymidine phospharamidite labelled at C6 with 13C has been synthesized, and incorporated into a synthetic oligonuckotide, d(CGCGT*T*GT*T*CGCG), which adopts a hairpin conformation. NMR relaxation measurements indicate that internal motion may be present in the loop region of the oligonucleotide. The relaxation behavior of a the C6 carbon in a model compound, N,N-1,3 dimethylthymine is examined in detail as a function of magnetic field strength to determine relative contributions of various mechanisms to the relaxation. The relaxation behaviour of the labelled carbons in the oligonucleotide is discussed in relation to these measurements.